
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Medico.ru2
Clinical Features and Outcomes of Childhood
Results From a National Population-Based Study
Piers E.F. Daubeney, MBBS; Alan W. Nugent, MBBS; Patty Chondros, MSc; John B. Carlin, PhD;
Steven D. Colan, MD; Michael Cheung, MB, ChB; Andrew M. Davis, MD; C.W. Chow, MD;
Robert G. Weintraub, MBBS; on behalf of the National Australian Childhood Cardiomyopathy Study
Background—Despite considerable mortality, population-based prognostic factors for childhood dilated cardiomyopathy
are lacking.
Methods and Results—A population-based cohort study was undertaken of all children in Australia who presented with
cardiomyopathy at age 0 to 10 years between January 1, 1987, and December 31, 1996. A single cardiologist analyzedall cardiac investigations, and a single pathologist analyzed histopathological material. There were 184 subjects withdilated cardiomyopathy. Positive viral identification or lymphocytic myocarditis was found in 30 (68.2%) of 44 caseswith available early histology and 8 of 9 cases presenting with sudden death. Freedom from death or transplantation was72% (95% CI, 65% to 78%) 1 year after presentation and 63% (95% CI, 55% to 70%) at 5 years. By proportionalhazards regression analysis, risk factors for death or transplantation comprised age ⬎5 years at presentation (hazard ratio5.6, 95% CI, 2.6 to 12.0), familial dilated cardiomyopathy (hazard ratio, 2.9; 95% CI, 1.5 to 5.6), lower initial fractionalshortening
z score (hazard ratio per
z-score unit, 0.75; 95% CI, 0.65 to 0.87), and failure to increase fractional shortening
z score during follow-up (hazard ratio per unit increase, 0.68; 95% CI, 0.58 to 0.79). At follow-up, 78 (44.6%) of 175cases diagnosed during life have no symptoms and are not taking any cardiac medication.
Conclusions—Early mortality is high in childhood dilated cardiomyopathy, but the clinical status of long-term survivors
is good. This population-based study identifies children at risk of adverse events.
(Circulation. 2006;114:2671-2678.)
Key Words cardiomyopathy 䡲 heart failure 䡲 myocarditis 䡲 pediatrics
Clinical Perspective p 2678
myopathy1,2 and is associated with considerable morbidity and
mortality.3–10 International registry data indicate that dilated cardio-
therapies.12,13 The present study examines the clinical char-
myopathy accounts for ⬎50% of all cardiac transplantations per-
acteristics and risk factors for death and transplantation
formed in patients between 1 and 10 years of age.11
among children with dilated cardiomyopathy enrolled in the
Reported outcomes for childhood dilated cardiomyopathy
National Australian Childhood Cardiomyopathy Study.
vary widely and have usually been based on institutional
reviews3,4,6–8 or limited geographic regions.9 Institutional
The National Australian Childhood Cardiomyopathy Study is a
reviews may not detect children who die soon after presen-
population-based cohort study of all children in Australia who
tation, and prognostic variables derived from population-
presented with cardiomyopathy at 0 to 10 years of age between
based studies are presently lacking. Some studies have
January 1, 1987, and December 31, 1996. Cases were recruited
examined outcomes only in relation to patient characteristics
during a series of site visits undertaken in 1997 to 2000 by the same3 investigators, who visited all 9 pediatric cardiac centers and an
at the time of initial evaluation. A better understanding of the
additional 12 hospitals caring for children with heart problems.
spectrum and outcomes of childhood dilated cardiomyopathy
Cases were also recruited from rural pediatricians, cardiac transplant
would facilitate patient care and permit evaluation of newer
centers, and cardiologists caring primarily for adults. In each
Received April 20, 2006; revision received September 17, 2006; accepted October 11, 2006.
From the Departments of Cardiology (P.E.F.D., A.W.N., M.C., A.M.D., R.G.W.) and Anatomic Pathology (C.W.C.), Royal Children's Hospital,
Melbourne, Australia; Clinical Epidemiology and Biostatistics Unit (P.C., J.B.C.), Murdoch Children's Research Institute, Melbourne, Australia;Departments of General Practice (P.C.) and Paediatrics (J.B.C., C.W.C.), University of Melbourne, Melbourne, Australia; and Department of Cardiology,Children's Hospital and Department of Pediatrics, Harvard Medical School, Boston, Mass (S.D.C.).
The online-only Data Supplement, which includes the medical institutions and physicians participating in the National Australian Childhood Cardio-
myopathy Study, is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.635128/DC1.
Guest Editor for this article was Robyn J. Barst, MD.
Correspondence to Dr Robert Weintraub, Department of Cardiology, Royal Children's Hospital, Flemington Rd, Parkville, VIC 3052, Australia. E-mail
2006 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org
December 12, 2006
location, study subjects were identified from multiple sources,
excluded 9 subjects whose initial manifestation was sudden death.
including cardiology and medical record databases and echocardi-
Important prognostic factors from the univariate analysis (
P⬍0.10)
ography logbooks. Children with cardiac dysfunction associated with
were included in a multivariable Cox proportional hazards model.
progressive neuromuscular disorders or inborn errors of metabolism
Lymphocytic myocarditis on endomyocardial biopsy was predictive
with multiple organ involvement were excluded. The methodology
of survival on univariate analysis but was not entered into the
and epidemiological findings have been described previously.1
multivariable model because cardiac histology was not available in
Ethics committee approval was obtained from each participating
all subjects. Because echocardiographic measurements were highly
interrelated, only fractional shortening
z score at presentation and
Cardiomyopathies were categorized according to the current
change in fractional shortening
z score on subsequent examinations
World Health Organization cardiomyopathy classification by a
were entered into the multivariable model (as continuous predictors,
single pediatric cardiologist after review of relevant investigations,
with the latter a time-dependent covariate defined by the most recent
including direct visualization and reinterpretation of all available
available echocardiographic value). Survival curves were plotted
cardiac imaging.14 The presence of congestive heart failure was
with Kaplan-Meier survival estimates, with accompanying 95%
based on signs and symptoms recorded by the attending physician.
Greenwood confidence bands.
The diagnostic criteria for dilated cardiomyopathy were (1) reduced
Follow-up data were available at least 2 years after presentation in
left ventricular systolic function on any form of cardiac imaging in
⬎90% of surviving subjects. The Wilcoxon rank-sum test was used
subjects with symptoms or a family history of dilated cardiomyop-
to compare the distribution of time from diagnosis between subjects
athy, (2) a measured left ventricular ejection fraction ⬍45% or a
with and without lymphocytic myocarditis in those with available
fractional shortening ⱕ20% in children without symptoms or a
myocardial histology. The Fisher exact test was used to examine the
positive family history, or (3) pathological evidence of dilated
association between inotropic support at presentation and the pres-
cardiomyopathy at autopsy. Although most subjects had left ventric-
ence or absence of lymphocytic myocarditis in the same subjects.
ular dilatation, this was not required for study inclusion, because
Analysis was undertaken with Stata software.19 Ninety-five per-
some subjects with rapidly progressive symptoms had normal left
cent CIs are given for estimated hazard ratios, and all reported
ventricular size at presentation. Subjects with lymphocytic myocar-
probability values are 2-sided.
ditis and associated left ventricular systolic dysfunction were in-
The authors had full access to the data and take full responsibility
cluded because their clinical and echocardiographic findings were
for the integrity of the data. All authors have read and agreed to the
frequently indistinguishable from those of other study subjects. A
manuscript as written.
single pediatric pathologist who was unaware of clinical patientdetails examined all available pathological specimens, includingcardiac histology.
Autopsy records were obtained from a computerized index kept by
The study population included all Australian children diag-
the Australian Bureau of Statistics that used the same diagnostic
nosed with dilated cardiomyopathy during the study period,
codes as for hospital records. In this way, information was obtained
as well as additional subjects identified at autopsy. Table 1
about subjects who had never had contact with a physician. Prospec-
summarizes the characteristics of the study population. The
tive clinical and echocardiographic follow-up was arranged forsurviving subjects, particularly those not seen within the preceding
majority of subjects had both left ventricular dilatation and
systolic dysfunction at diagnosis.
Specifically designed data forms were used to ascertain and record
uniform clinical and epidemiological retrospective information for
Presenting Symptoms and Initial Therapy
each enrolled subject from all available hospital and outpatient case
Congestive heart failure was the presenting symptom in 165
records, including the results of all relevant investigations. Datarecorded during prospective follow-up were recorded in the same
(89.7%) of 184 patients, sudden death in 9 (4.9%), and other
way. Prognostic factors sought included clinical features at presen-
symptoms (exercise intolerance or arrhythmias) in 4 (2.2%),
tation and results of relevant investigations (see Appendix in the
whereas 6 cases (3.3%) were detected solely on routine
online Data Supplement). The earliest available ECG within 7 days
screening of family members. The median age of the 9 cases
of presentation was read by a single observer, and measurementswere converted to age-appropriate
z scores.15 Serial echocardio-
whose initial symptom was sudden death was 2 months
graphic measurements of left ventricular dimensions, wall thickness,
(range 8 days to 11 months). Of the 175 patients who were
and fractional shortening (in those without regional wall-motion
diagnosed during life, 154 (88%) were hospitalized, and 79
abnormalities), were expressed as
z scores based on body surface
(45.1%) were admitted to an intensive care unit. Established
area (or age, in the case of fractional shortening).16,17 These
renal failure that required dialysis or specific supportive
echocardiographic parameters were selected because they wereroutinely quantified in all subjects who underwent echocardiography
measures was present in 18 patients (10.3%) at diagnosis, and
and were usually available at presentation, after 3, 6, 12, and 24
rhabdomyolysis was present in 2 (1.1%). Assisted mechanical
months, and at latest follow-up. Among subjects with serial echo-
ventilation was administered in 62 patients (35.4%) and
cardiograms available from the time of presentation, the change in
inotropic support in 70 (40%). The frequency with which
fractional shortening (and fractional shortening
z score) at eachsubsequent occasion of measurement was calculated by subtracting
inotropic support was administered at presentation was sim-
the initial fractional shortening (fractional shortening
z score) from
ilar among children with lymphocytic myocarditis (9 of 25
the subsequent value.
cases, 36%) compared with those with nonspecific histolog-
Familial cardiomyopathy was considered to be present when there
ical findings (18 of 45 cases, 40%;
P⫽0.80). Long-term
was an affected first- or second-degree relative with primary dilatedcardiomyopathy identified from the case notes or from prospective
medical therapy for congestive heart failure, including diuret-
screening of other family members. Definite lymphocytic myocar-
ics, digoxin, an afterload-reducing agent (usually an angio-
ditis was classified according to the Dallas criteria.18 Routine genetic
tensin-converting enzyme inhibitor), an aldosterone antago-
testing for mutations known to cause dilated cardiomyopathy was not
nist, or a -blocker, was initiated at some point in 134
available during the study period.
subjects (76.6%), and systemic anticoagulation was adminis-
tered in 24 (13.7%). Immune modulating therapy (cyclospor-
Standard methods of survival analysis were used for the combined
ine, steroids, or ␥-globulin) was used in 11 of 13 children
end point of death or transplantation. Analysis of prognostic factors
with lymphocytic myocarditis on endomyocardial biopsy.
Daubeney et al
Childhood Dilated Cardiomyopathy
Demographic Characteristics of the Patients
logical findings for lymphocytic myocarditis (Table 2;
P⫽0.009). Although viral identification from endomyocardi-al biopsy was not routinely undertaken during the study
period, 8 cases had both lymphocytic myocarditis and posi-
Proportion of total cases in NACCS (n⫽314)
tive viral identification from at least 1 source. When micro-
Male/female, n (%)
biological and histological investigations were considered
Age at presentation, n (%)
together, a potential viral contribution was identified in 58
(31.5%) of 184 cases, including 30 (68.2%) of 44 cases who
⬎4 wk and ⱕ1 y
underwent cardiac histological examination within 7 days of
⬎1 y and ⱕ5 y
presentation and 6 (66.7%) of 9 whose initial manifestation
was sudden death.
Familial cardiomyopathy, n (%)
Familial Cardiomyopathy, Metabolic Conditions, and
Lymphocytic myocarditis, No. of cases with
available histology (%)
Familial dilated cardiomyopathy was identified in 27 subjects
Presence of metabolic disease, n (%)
(14.7%), 8 of whom did not have symptoms at diagnosis.
Death or transplantation, n (%)
Three (11.1%) of 27 children with familial cardiomyopathy
Years of follow-up from presentation for all
showed genetic anticipation, with a prior affected family
patients (n⫽175*)
member having onset of cardiomyopathy during adult life. A
metabolic or mitochondrial disease (Barth syndrome, carni-
Interquartile range
tine transport defect, fatty acid oxidation defect, or respira-
Years of follow-up from presentation for surviving
tory chain complex deficiency) with predominant cardiac
patients (n⫽109)
manifestations was diagnosed in 12 (8.9%) of 135 cases who
underwent at least 1 metabolic investigation, 8 of whompresented at age
Interquartile range
⬍12 months. Parental consanguinity was
present in 14 (8.8%) of 160 cases in which this variable could
LV end-systolic dimension
z score at presentation
be ascertained, including 3 with familial cardiomyopathy.
Interquartile range
A total of 75 patients (40.8%) have died or undergone
LV end-diastolic
z score at presentation (N⫽150†)
transplantation. These included 9 subjects who presented with
sudden death and 24 who died during their initial hospital-
Interquartile range
ization at a median of 2 (range, 0 to 18) days after
Fractional shortening at presentation (n⫽150†)
At latest follow-up, 78 of 184 subjects are taking no regular
Interquartile range
medication, whereas 31 (16.8%) are receiving long-termmedical therapy. Of the 103 patients with known symptom-
Fractional shortening
z score at presentation(n⫽150†)
atic status, 94 (91.3%) are without cardiac symptoms.
Interquartile range
⫺12.1 to ⫺8.7
Actuarial freedom from death or transplantation was 72%
NACCS indicates National Australian Childhood Cardiomyopathy Study; LV,
(95% CI, 65% to 78%) 1 year after presentation and 63%
left ventricular.
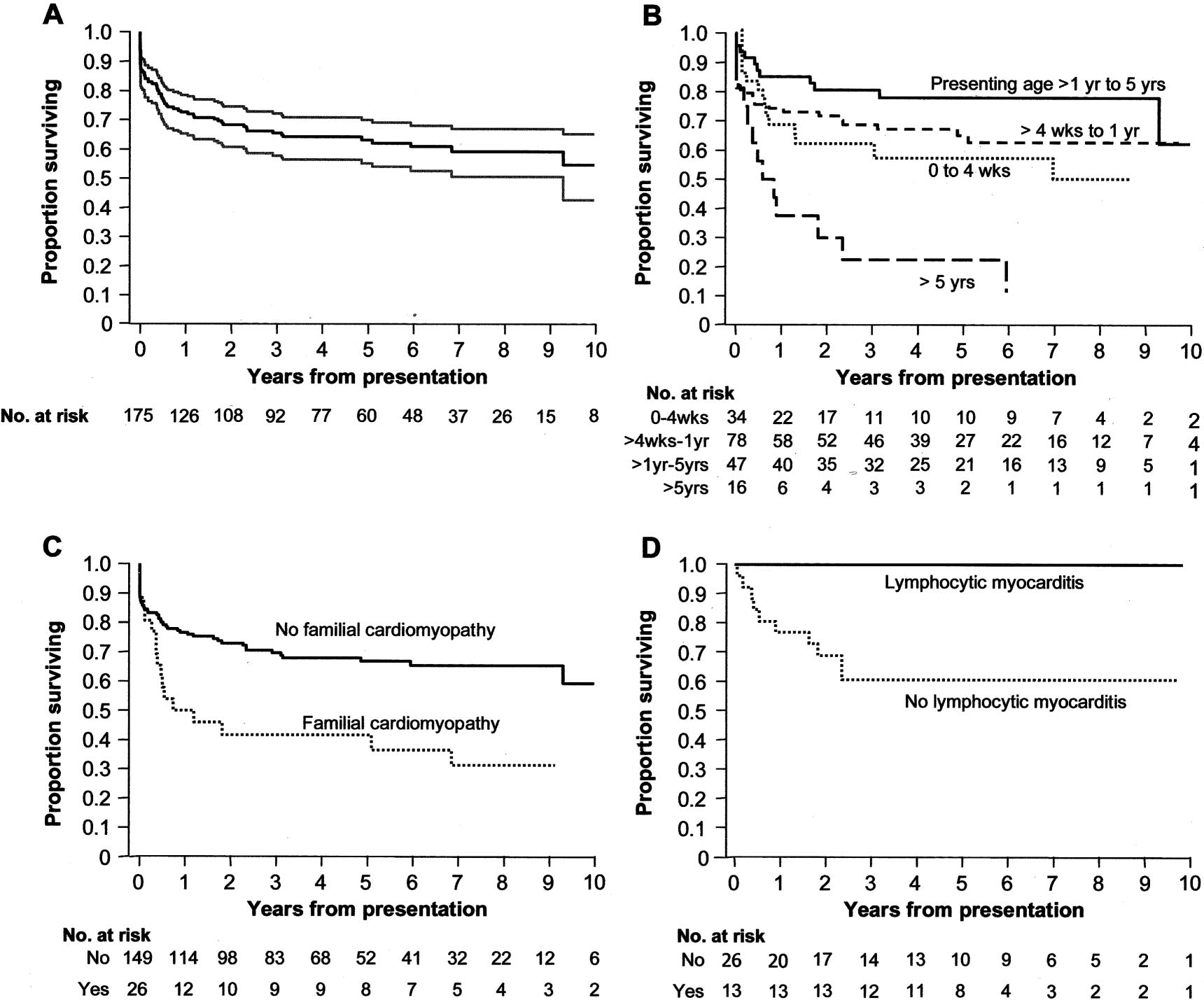
(95% CI, 55% to 70%) at 5 years (Figure, panel A). Risk
n denotes the total number of patients with any form of cardiomyopathy
factors showing at least weak evidence (
P⬍0.1) of associa-
over the 10-year ascertainment period in Australia.
tion with risk of death or transplantation are shown in Table
*Total excludes 9 subjects whose first manifestation was sudden death.
†Twenty-five subjects did not have a suitable echocardiogram for analysis at
3. There was a clear difference in survival according to age at
presentation (
P⫽0.001), mainly due to much worse outcomesamong children aged ⬎5 years at diagnosis (Table 3; Figure,
panel B). There was also some indication that survival was
Viral Identification and Lymphocytic Myocarditis
lower among infants aged 0 to 1 month and those aged 1 to
In 41 (22.3%) of 184 subjects, a potentially cardiotoxic virus
12 months than among those aged 12 months to 5 years at
(most commonly coxsackievirus or adenovirus) was identi-
diagnosis (post hoc 3-group comparison
P⫽0.11). Given the
fied from urine, stools, or upper-airway secretions at presen-
weak evidence for differences between the 3 youngest age
tation with polymerase chain reaction or viral culture. Lym-
groups, presenting age was dichotomized at 5 years for entry
phocytic myocarditis was present in 25 (35.7%) of 70 cases
in the multivariable proportional hazards model. In this
with available cardiac histology from any source (endomyo-
model, factors that showed evidence of independent predic-
cardial biopsy, explantation, or autopsy). There was an
tive associations included age ⬎5 years at presentation
inverse relationship between the time from diagnosis to
(Figure, panel B), familial dilated cardiomyopathy (Figure,
histological examination and the presence of positive histo-
panel C), a lower fractional shortening
z score at presentation,
December 12, 2006
Frequency of Lymphocytic Myocarditis Among Subjects With Available
Cardiac Histology, According to Time Between Diagnosis and Examination of
Histological Findings (nⴝ
70)
Time From Presentation
⬎1–4 Weeks ⬎4–8 Weeks ⬎8 Weeks Total
Lymphocytic myocarditis
Nonspecific histological findings
P⫽0.009, Wilcoxon rank-sum test.
and failure to increase fractional shortening
z score from
number of subjects who died before recognition of their
condition was substantially underestimated. In particular, the
At 2 years after presentation, 24 (13.7%) of the 175
number of cases diagnosed from autopsy after sudden unex-
patients who were diagnosed during life had a measured
pected death has highlighted the significant early mortality in
fractional shortening below 20%. Outcomes for this group
childhood dilated cardiomyopathy.
were poor. Eleven (45.8%) of the 24 died or underwenttransplantation, and all of the remaining subjects continue to
Symptoms and Causes
have impaired left ventricular function.
Symptoms were present in most subjects at presentation andwere usually severe. Congestive heart failure was the initial
Lymphocytic Myocarditis
Of 25 children with lymphocytic myocarditis, 12 (48%) were
symptom in almost 90% of patients, half of whom were
diagnosed at autopsy. Six (24%) of these presented with
admitted to an intensive care unit. Sudden death was the first
sudden death, and the other 6 (24%) died within 3 days of
manifestation of dilated cardiomyopathy in nearly 5%, and a
presentation. The remaining cases were diagnosed from
further 13% died during their initial hospitalization.
endomyocardial biopsy. Among 39 children who underwent
Lymphocytic myocarditis among adults with left ventric-
endomyocardial biopsy, survival among the 13 cases with
ular dysfunction occurs in ⬇10% of cases22 and may be due
lymphocytic myocarditis was significantly better than among
to a variety of viral and autoimmune causes.23 In children
the 26 who had nonspecific histological findings (Table 2;
with dilated cardiomyopathy, lymphocytic myocarditis is
Figure, panel D). At latest follow-up in children with lym-
found more frequently5 and more commonly reflects a viral
phocytic myocarditis diagnosed from endomyocardial biopsy,
origin.24–26 The results of viral identification from tracheal
the mean (interquartile range) left ventricular end-diastolic
aspirate by polymerase chain reaction have been shown to
dimension and fractional shortening
z scores were 0.4
correlate well with those obtained from myocardium and the
(⫺0.65, 0.7) and ⫺0.5 (⫺1.2, 0.8), respectively.
lower respiratory tract.25 In the present study, positive viralidentification or lymphocytic myocarditis was present in 68%
of subjects with early available myocardial histology, includ-
Population-based studies have provided unique insights into
ing 6 of 9 subjects presenting with sudden death. The
disease severity and outcomes among adults with cardiomy-
diagnosis of postviral cardiomyopathy remains problematic,
opathy.20 The National Australian Childhood Cardiomyopa-
and these findings are open to a number of interpretations.
thy study is the largest population-based study of childhood
Low ascertainment of viral illness may be due to collection of
cardiomyopathy1 and therefore provides useful reference
specimens well after the viremic phase of the initial illness,
data. Consistency of case classification was maintained by
low utilization of early endomyocardial biopsy, and lack of
having the same observers examine all available cardiac
direct viral testing on myocardial samples, whereas overas-
investigations and histopathological material. Although the
certainment may be due to spurious association with systemic
number of cases with mild unrecognized dilated cardiomy-
viral illnesses. The present study demonstrates a potential
opathy remains unknown, case ascertainment for children
viral contribution in a high proportion of cases, however. This
already diagnosed is likely to have been effectively complete
unexpectedly high prevalence may reflect the inclusion of
for the following reason: The majority of children who arediagnosed with dilated cardiomyopathy come to early medi-
autopsy cases and subjects who would otherwise not have
cal attention because of severe symptoms.21 Centralization of
come to attention in an institutional review. The inverse
tertiary services within Australia enabled cases to be recruited
relation between the prevalence of myocarditis and increasing
from multiple sources, which included all pediatric cardiac
time since presentation is consistent with animal models in
centers and cardiologists, as well as from rural pediatricians
which myocardial inflammation disappears within 6 weeks of
and physicians caring primarily for adults. Finally, the inci-
viral inoculation.27
dence of dilated cardiomyopathy in the present study was
Other potential causes, such as familial dilated cardiomy-
similar to that of a recent North American study,1,2 which
opathy, a metabolic disease, and parental consanguinity (as a
indicates that case ascertainment was likely to be high.
marker for a recessively inherited condition), were each
Because Australian law requires autopsies to be performed in
documented in 8.8% to 14.7% of study subjects. Familial
cases of sudden or unexplained death, it is unlikely that the
cardiomyopathy and mitochondrial diseases may well have
Daubeney et al
Childhood Dilated Cardiomyopathy
Survival to death or transplantation from time of presentation: overall, with 95% CI (A); with patients grouped by age at presentation(B); with patients grouped by presence or absence of familial cardiomyopathy (C); and with patients grouped by presence or absenceof lymphocytic myocarditis on endomyocardial biopsy (D).
been underrecognized, because not all subjects and families
ed.30 Few pediatric studies have examined late survival in
were systematically screened.28
relation to serial assessment of left ventricular function.
In the present study, independent risk factors for death or
transplantation comprised older age at presentation, familial
Reported outcomes and prognostic factors for childhood
dilated cardiomyopathy, lower initial fractional shortening z
dilated cardiomyopathy have varied considerably. Five-year
score, and failure to increase fractional shortening z score
survival rates of 20% to 84% have been described from
during follow-up. Younger patients had a nonsignificant trend
institutional reviews.3,6,8,10 In 1 study of 24 children pres-
to worse survival, which suggests the possibility of a bimodal
enting at ⬍2 years of age,4 absence of lymphocytic myocar-
age distribution for poor outcomes in childhood dilated
ditis, a spherical left ventricular shape, and more depressed
cardiomyopathy. The impact of each established risk factor
left ventricular function at presentation were associated with
was considerable. For example, an increase in fractional
worse outcomes. Another review of 25 children aged ⬎2
shortening z score over the baseline value measured at
years at presentation reported a 3-year survival of only 20%.6
presentation was associated with a 32% reduction in hazard
Some studies have found no relation between late survival
ratio per unit z score. Improvement in left ventricular function
and age3,7 or severity of cardiac dysfunction3,6–8 at presenta-
among surviving subjects may have been due to supportive
tion. Other proposed adverse risk factors include the magni-
medical therapy and, in some cases, resolution of postviral
tude of diastolic dysfunction at cardiac catheterization3 or by
cardiomyopathy. The clinical status of long-term survivors
echocardiography (in adults).29 A single previous national
was good, with nearly half of all study subjects having no
study30 of 62 cases who were diagnosed between 1980 and
symptoms and no longer receiving cardiological medications.
1991 reported that right ventricular failure at presentation and
Despite a high initial mortality, subjects with lymphocytic
requirement for anticoagulant therapy were associated with
myocarditis diagnosed during life had a better survival than
worse outcomes. The study selection criteria differed from
those with nonspecific histological findings. This may reflect
the present study, however, in that subjects with myocarditis
the rapid evolution of pediatric lymphocytic myocarditis,
and those with self-limiting cardiomyopathies were exclud-
selection of less critically ill patients for biopsy, a more
December 12, 2006
Survival Analysis of Predictors of Death or Transplantation (nⴝ175)
Multivariable Survival Analysis
Univariable Survival Analysis
Age at presentation (4 groups)
⬎4 wk and ⱕ1 y
0.73 (0.39–1.4)
⬎1 y and ⱕ5 y
0.44 (0.20–0.95)
2.26 (1.07–4.8)
Age at presentation (2 groups) ⬎5 y
3.27 (1.77–6.0)
Familial cardiomyopathy
2.35 (1.35–4.1)
Biopsy myocarditis*
QRS duration z score
1.38 (1.02–1.9)
Fractional shortening at presentation‡
0.92 (0.87–0.97)
Change in fractional shortening from presentation‡
0.83 (0.78–0.89)
Fractional shortening z score at presentation‡
0.85 (0.75–0.96)
0.75 (0.65–0.87)
Change in fractional shortening z score from presentation‡
0.66 (0.57–0.77)
0.68 (0.58–0.79)
Excludes 9 subjects whose first manifestation was sudden death. Measured QRS duration was from the earliest available ECG. For the 3 youngest
age groups, presenting age was dichotomized at 5 years for entry in the multivariable proportional hazards regression model.
Only variables that achieved a P value ⬍0.10 are included in the table; other variables examined are listed in the Appendix.
*The finding of myocarditis on biopsy was significantly predictive of survival, but a hazard ratio could not be calculated because all patients with
this characteristic were free from death or transplant; P value calculated with log-rank test.
†P values from Wald tests in Cox proportional hazards regression, with 3 degrees of freedom in case of 4-group comparison and 1 degree of
freedom otherwise.
‡Per unit (percent fractional shortening or unit z score).
favorable natural history of this condition, or the benefits of
tation and the lack of sufficient young donors justify the
the various therapies employed. Among adults with lympho-
continuing search for better medical therapy.
cytic myocarditis, fulminant onset is associated with im-proved outcomes,31 although there is no evidence that immu-
nosuppressive therapy modifies the natural history.22
The Dallas criteria are of limited utility in the diagnosis of
However, there exist numerous reports of favorable outcomes
postviral cardiomyopathy and are subject to considerable
after treatment among both children5,32–34 and subgroups of
interobserver variability.38 The latter was minimized by
adults with lymphocytic myocarditis.23,31,35 A separate trial of
having a single pediatric pathologist examine all available
immune-modulating therapy in children appears warranted to
myocardial histology. Retrospective data collection, variable
address this issue.
diagnostic protocols, and limitations in existing knowledgealso restricted the proportion of study subjects with cardio-
myopathy of known origin. By comparison with other pedi-
The present study took place during a period of rapidly
atric studies, however, the proportion of subjects with a
changing medical therapy for subjects with dilated cardiomy-
known or probable cause remains high. Routine genetic
opathy. The data supporting the efficacy of medical therapy
testing for dilated cardiomyopathy was not available during
in children with dilated cardiomyopathy are less robust than
the study period, and the results of the present study cannot be
in adult subjects, and the present study was not designed to
extrapolated to children who have not yet developed left
draw conclusions about the benefits of any specific treatment.
ventricular dysfunction. A more global measure of left
In adult patients with left ventricular dysfunction, carvedilol
ventricular systolic function and routine measurement of
reduces mortality36 and modifies prognostic factors.37 Retro-
diastolic parameters may further increase the predictive value
spective studies in children suggest that -blockers improve
of echocardiography. These were not serially available on all
ventricular performance,12 and a prospective multicenter trial
subjects during the study period. The observation that infants
is presently under way. Although mortality and requirement
may also be at increased risk for worse outcome may be
for eventual cardiac transplantation may be reduced by
confirmed with a larger study cohort.
aggressive use of newer medical therapies in children, theirimpact in the youngest of patients has yet to be defined. The
present study identifies children at greatest risk for death or
Congestive heart failure is severe among children with dilated
transplantation, who might therefore benefit most from effec-
cardiomyopathy, and lymphocytic myocarditis is an impor-
tive early treatment.10 The long-term limitations of transplan-
tant cause. Although early mortality is high, the clinical status
Daubeney et al
Childhood Dilated Cardiomyopathy
of long-term survivors is good. This population-based study
eration of Cardiology Task Force on the Definition and Classification of
identifies children with dilated cardiomyopathy who are at
Cardiomyopathies. Circulation. 1996;93:841– 842.
15. Nugent AW, Davis AM, Kleinert S, Wilkinson JL, Weintraub RG.
risk for adverse events.
Clinical, electrocardiographic, and histologic correlations in childrenwith dilated cardiomyopathy. J Heart Lung Transplant. 2001;20:1152–1157.
16. Sluysmans T, Colan SD. Theoretical and empirical derivation of cardio-
The authors are indebted to Ingrid King for assistance with database
vascular allometric relationships in children. J Appl Physiol. 2005;99:
17. Colan SD, Parness IA, Spevak PJ, Sanders SP. Developmental modu-
lation of myocardial mechanics: age- and growth-related alterations in
Sources of Funding
afterload and contractility. J Am Coll Cardiol. 1992;19:619 – 629.
This study was supported by grants from the Murdoch Children's
18. Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio
Research Institute (grant 98001), the National Heart Foundation of
JJ Jr, Olsen EG, Schoen FJ. Myocarditis. A histopathologic definition and
Australia (grants G98M0159 and G05M2151), and the Australia and
classification. Am J Cardiovasc Pathology. 1987;1:3–14.
New Zealand Children's Heart Research Centre.
19. Stata Statistical Software [computer program]. Release 8.0. College
Station, Tex: StataCorp LC; 2003.
20. Spirito P, Chiarella F, Carratino L, Berisso MZ, Bellotti P, Vecchio C.
Clinical course and prognosis of hypertrophic cardiomyopathy in an
outpatient population. N Engl J Med. 1989;320:749 –755.
21. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL,
Tikanoja T, Paavilainen T, Simell O. Epidemiology of idiopathic cardio-
myopathies in children and adolescents: a nationwide study in Finland.
Am J Epidemiol. 1997;146:385–393.
1. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson
22. Mason JW, O'Connell JB, Herskowitz A, Rose NR, McManus BM,
LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG;
Billingham ME, Moon TE; the Myocarditis Treatment Trial Investigators.
National Australian Childhood Cardiomyopathy Study. The epidemiolo-
A clinical trial of immunosuppressive therapy for myocarditis. N Engl
gy of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:
J Med. 1995;333:269 –275.
1639 –1646.
23. Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A.
2. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF,
Immunosuppressive therapy for active lymphocytic myocarditis: viro-
Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The
logical and immunologic profile of responders versus nonresponders.
incidence of pediatric cardiomyopathy in two regions of the United
States. N Engl J Med. 2003;348:1647–1655.
. 2003;107:857– 863.
3. Lewis AB, Chabot M. Outcome of infants and children with dilated
24. Martin AB, Webber S, Fricker FJ, Jaffe R, Demmler G, Kearney D,
cardiomyopathy. Am J Cardiol. 1991;68:365–369.
Zhang YH, Bodurtha J, Gelb B, Ni J, Bricker T, Towbin JA. Acute
4. Matitiau A, Perez-Atayde A, Sanders SP, Sluysmans T, Parness IA,
myocarditis: rapid diagnosis by PCR in children. Circulation. 1994;90:
Spevak PJ, Colan SD. Infantile dilated cardiomyopathy: relation of
outcome to left ventricular mechanics, hemodynamics, and histology at
25. Akhtar N, Ni J, Stromberg D, Rosenthal GL, Bowles NE, Towbin JA.
the time of presentation. Circulation. 1994;90:1310 –1318.
Tracheal aspirate as a substrate for polymerase chain reaction detection of
5. Kleinert S, Weintraub RG, Wilkinson JL, Chow CW. Myocarditis in
viral genome in childhood pneumonia and myocarditis. Circulation.
children with dilated cardiomyopathy: incidence and outcome after dual
therapy immunosuppression. J Heart Lung Transplant. 1997;16:
26. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP,
1248 –1254.
McCarthy R, Hare J, Bricker JT, Bowles KR, Towbin JA. Detection of
6. Akagi T, Benson LN, Lightfoot NE, Chin K, Wilson G, Freedom RM.
viruses in myocardial tissues by polymerase chain reaction: evidence of
Natural history of dilated cardiomyopathy in children. Am Heart J.
adenovirus as a common cause of myocarditis in children and adults.
J Am Coll Cardiol. 2003;42:466 – 472.
7. Wiles HB, McArthur PD, Taylor AB, Gillette PC, Fyfe DA, Matthews JP,
27. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflam-
Shelton LW. Prognostic features of children with idiopathic dilated car-
mation and cell death: learning from the past for the future. Circulation.
diomyopathy. Am J Cardiol. 1991;68:1372–1376.
8. Friedman RA, Moak JP, Garson A Jr. Clinical course of idiopathic dilated
28. Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna
cardiomyopathy in children. J Am Coll Cardiol. 1991;18:152–156.
WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common
9. Venugopalan P, Houston AB, Agarwal AK. The outcome of idiopathic
in asymptomatic relatives and may represent early disease. J Am Coll
dilated cardiomyopathy and myocarditis in children from the west of
Scotland. Int J Cardiol. 2001;78:135–141.
29. Pinamonti B, Zecchin M, Di Lenarda A, Gregori D, Sinagra G, Camerini
10. Tsirka AE, Trinkaus K, Chen SC, Lipshultz SE, Towbin JA, Colan SD,
F. Persistence of restrictive left ventricular filling pattern in dilated
Exil V, Strauss AW, Canter CE. Improved outcomes of pediatric dilated
cardiomyopathy: an ominous prognostic sign. J Am Coll Cardiol. 1997;
cardiomyopathy with utilization of heart transplantation. J Am Coll
29:604 – 612.
30. Arola A, Tuominen J, Ruuskanen O, Jokinen E. Idiopathic dilated car-
11. Boucek MM, Edwards LB, Keck BM, Trulock EP, Taylor DO, Hertz MI.
diomyopathy in children: prognostic indicators and outcome. Pediatrics.
Registry of the International Society for Heart and Lung Transplantation:
eighth official pediatric report–2005. J Heart Lung Transplant. 2005;24:
31. McCarthy RE III, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK,
Hare JM, Baughman KL. Long-term outcome of fulminant myocarditis as
12. Bruns LA, Chrisant MK, Lamour JM, Shaddy RE, Pahl E, Blume ED,
compared with acute (nonfulminant) myocarditis. N Engl J Med. 2000;
Hallowell S, Addonizio LJ, Canter CE. Carvedilol as therapy in pediatric
342:690 – 695.
heart failure: an initial multicenter experience. J Pediatr. 2001;138:
32. Lee KJ, McCrindle BW, Bohn DJ, Wilson GJ, Taylor GP, Freedom RM,
Smallhorn JF, Benson LN. Clinical outcomes of acute myocarditis in
13. Follath F, Cleland JG, Just H, Papp JG, Scholz H, Peuhkurinen K, Harjola
childhood. Heart. 1999;82:226 –233.
VP, Mitrovic V, Abdalla M, Sandell EP, Lehtonen L, Steering C; Inves-
33. Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL, Takahashi M,
tigators of the Levosimendan Infusion versus Dobutamine Study.
Baker AL, Perez-Atayde AR, Newburger JW. Gamma-globulin treatment
Efficacy and safety of intravenous levosimendan compared with dobuta-
of acute myocarditis in the pediatric population. Circulation. 1994;89:
mine in severe low-output heart failure (the LIDO study): a randomised
double-blind trial. Lancet. 2002;360:196 –202.
34. Camargo PR, Snitcowsky R, da Luz PL, Mazzieri R, Higuchi ML, Rati
14. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell
M, Stolf N, Ebaid M, Pileggi F. Favorable effects of immunosuppressive
J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report
therapy in children with dilated cardiomyopathy and active myocarditis.
of the 1995 World Health Organization/International Society and Fed-
Pediatr Cardiol. 1995;16:61– 68.
December 12, 2006
35. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G,
Effect of carvedilol on survival in severe chronic heart failure. N Engl
Wilczewski P, Niklewski T, Zembala M, Polonski L, Rozek MM, Wod-
J Med. 2001;344:1651–1658.
niecki J. Randomized, placebo-controlled study for immunosuppressive
37. Zugck C, Haunstetter A, Kruger C, Kell R, Schellberg D, Kubler W,
treatment of inflammatory dilated cardiomyopathy: two-year follow-up
Haass M. Impact of beta-blocker treatment on the prognostic value of
results. Circulation. 2001;104:39 – 45.
currently used risk predictors in congestive heart failure. J Am Coll
36. Packer M, Coats AJ, Fowler MB, Katus HA, Krum H, Mohacsi P,
Rouleau JL, Tendera M, Castaigne A, Roecker EB, Schultz MK, DeMets
38. Baughman KL. Diagnosis of myocarditis: death of Dallas criteria. Cir-
DL; Carvedilol Prospective Randomized Cumulative Survival Study G.
Pediatric cardiomyopathies are characterized by heterogenous origins with varied outcomes. Childhood dilated cardiomy-opathy is most common during the first year of life and is associated with significant morbidity and mortality. Mostavailable information is from institutional reviews, which may not detect children who die early and those who do notrequire hospitalization. Population-based studies of cardiomyopathy in adults have provided valuable insights into diseaseseverity and outcomes. The National Australian Childhood Cardiomyopathy study is a longitudinal cohort study of allchildren in Australia aged 0 to 10 years who were diagnosed with cardiomyopathy between 1987 and 1996. There were184 subjects with dilated cardiomyopathy. At presentation, 90% of cases had signs and symptoms of congestive heartfailure, and sudden death was the presenting symptom in 4%. Familial cardiomyopathy was identified in 14.7% of subjects,a metabolic or mitochondrial disease in 8.9%, and parental consanguinity, consistent with autosomal recessive inheritance,in 8.8% of cases. A potential viral contribution (lymphocytic myocarditis or positive viral identification) was identified in68.2% of case subjects who underwent early cardiac histological examination. By multivariate analysis, independent riskfactors for death or cardiac transplantation included age ⬎5 years at presentation, familial dilated cardiomyopathy, a lowerfractional shortening z score at presentation, and failure to increase fractional shortening z score from presentation. At latestfollow-up, 78 of 109 surviving cases had no symptoms and were not taking any cardiac medication. Early mortality is highin childhood dilated cardiomyopathy, and the clinical status of long-term survivors is good. The present population-basedstudy identifies children at risk of adverse outcomes.
Source: http://www.medico.ru/arhiv/Dilated%20Cardiomyopathy.pdf
Strategies to Prevent Ventilator-Associated Pneumonia in Acute Care Hospitals: 2014 Update Author(s): Michael Klompas, MD, MPH; Richard Branson, MSc, RRT; Eric C. Eichenwald, MD; Linda R. Greene, RN, MPS, CIC; Michael D. Howell, MD, MPH; Grace Lee, MD; Shelley S. Magill, MD, PhD; Lisa L. Maragakis, MD, MPH; Gregory P. Priebe, MD; Kathleen Speck, MPH; Deborah S. Yokoe, MD, MPH; Sean M. Berenholtz, MD, MHS
DIABETES MELLITUS Understanding Type 1 and Type 2 Diabetes and Disease Progression Buge Apampa PhD MRPharmS Some Questions to start off! 1. How many people are expected to have diabetes in the UK by 2025? [5m] 2. What is the estimated hourly cost of diabetes to 3. How many diabetic patients are dying avoidably each year? [24,000]


