
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Szuperantigének és tcr vß specificitásuk
AUTOIMMUNITY, AUTOIMMUNE
DISEASES
Prof dr Gergely Péter
Semmelweis University
Central Laboratory of Immunology
The etiology of autoimmune diseases is unknown.
They are diseases of variable clinical picture and
prognosis, their common feature: autoreactivity
(autoreactive T cells, autoantibodies) plays a pivotal role in
their pathogenesis.
Features:
•
Variable clinical picture and prognosis, even within one
disease (spontaneous remissions)
•
In the majority: Female predominance (e.g., in SLE 9:1)
•
They frequently appear as „overlap" e.g. Sjögren's
syndrome, mixed connective tissue disease (MCTD).
The key element in the pathogenesis: loss of
autotolerance.
The mechanism of autotolerance:
•
central tolerance (passive; occurs in the thymus, during
early stage of ontogenesis)
•
peripheral tolerance (mainly active processes, during
entire life)
Autoimmunity:
(loss of autotolerance)
causes:
1) intrinsic: genetic (MHC, TCR, IG, cytokine, etc)
2) extrinsic:
hormonal (female predominance)
drugs, UV, etc. (rare)
infections
bacteria (cross reactivity, super-
antigens)
viruses (cross reactivity, polyclonal
B cell stimulation, disturbed immune
regulation)
EXTRINSIC FACTORS:
Role of infections in triggering auto-
immune diseases
1) Cross-reacting antigens or molecular mimicry
2) polyclonal T & B cell activation
(e.g. superantigenes)
3) tissue injury – loss of barrier function
4) bystander activation
5) epitope spreading
INTRINSIC FACTORS:
The role of MHC in disease susceptibilty
HLA antigen
Ankylosing spondylitis
Reiter's syndrome
Rheumatoid arthritis
DR4 (B1*0401/04)
Juvenile RA (JIA)
Multiple sclerosis
INTRINSIC FACTORS: Genetic background of SLE
Gene
Chromosome
Importance
Fc receptor:
Cytokine:
HLA-DR2/DR3 6p21
Complement:
10q11.2-q21
Apoptosis:
PATHOGENIC PROCESSES IN AUTOIMMUNE DISEASES:
a) autoantibodies:
direct cytotoxicity - complement/lysis -
enhanced phagocytosis
inhibition of function - myasthenia
stimulation - TSI
cell activation - ANCA
antiphospholipid antibodies - thrombosis
other - intracellular antigens (ANA)
b) IC disease – key feature of SLE
c) T cells
perforin - necrosis
granzyme B - apoptosis
both Th1 & Th2 cytokines
HUMAN AUTOIMMUNE DISEASES
1. Systemic
systemic lupus erythematosus (SLE)
antiphospholipid syndrome (APS)
rheumatoid arthritis (RA)
Sjögren's syndrome (SS)
scleroderma / mixed connective tissue disease (MCTD)
myositis group
undifferentiated (UCTD and overlap)
chronic graft versus host disease (GVHD)
2. Organ-specific
e.g. Hashimoto, AIHA, ITP, etc.
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
Definition: The clinical picture of SLE is variable. The most
common form of the disease: young female with non-erosive
arthritis, (low grade) fever, elevated ESR, leucopenia (with or
without skin and kidney involvement).
Epidemiology: Incidence: 0.7/100,000, prevalence 23-50/100,000. In
Hungary the expected number of SLE cases may be between 2500
and 5000. Female dominance (male: female ratio: 1:9). Familial
aggregation is known.
Clinical features of SLE
Clinical picture: The well known butterfly (malar) rash is relatively
uncommon, but the diagnosis is in most cases easy, if we base it on
the criteria of American Rheumatism Association (ARA), now called
American College of Rheumatology (ACR).
General symptoms: fever, lymphadenomegaly, weight loss are always
present, their severity depends on the intensity of the disease.
At first sight, such patient seem to be suffering of an infectious
disease.
Joints: are frequently involved, moderate swelling and morning
stiffness are the dominant features. Unlike to RA, no erosions and
deformity is seen. Subluxation and Jaccoud type arthropathy are
rare forms of SLE.
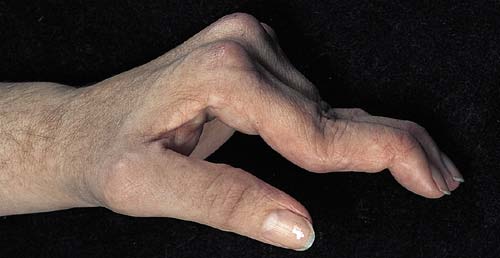
Jaccoud arthropathy in SLE
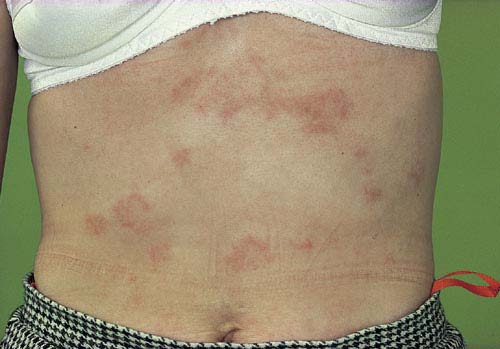
Skin involvement is frequent. Beside butterfly and discoid rash,
chronic (IC-mediated) urticaria, livedo reticularis, skin vasculitis, biphasic Raynaud's phenomenon, hair thickening and alopecia areata may be present.
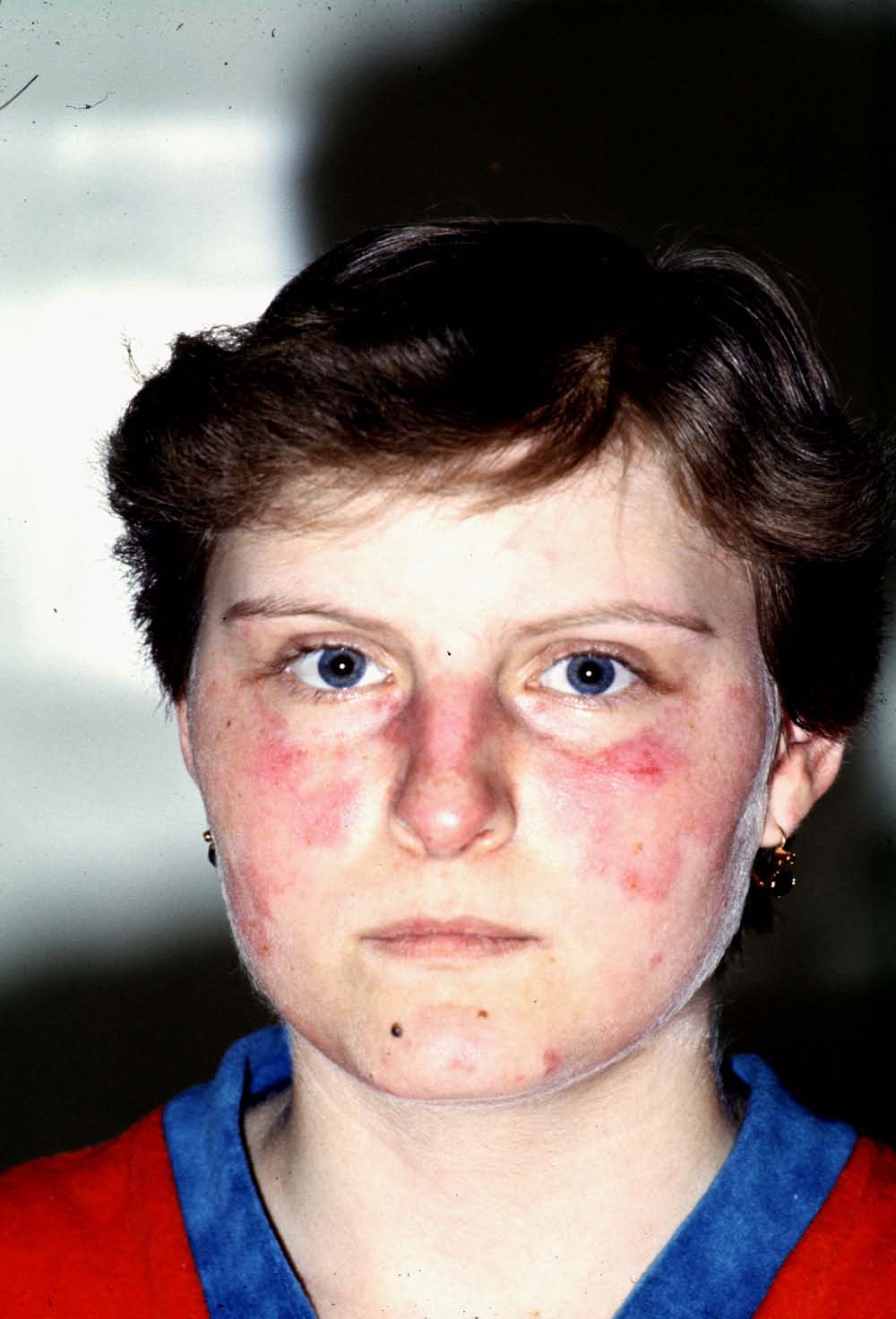
Malar rash in
SLE
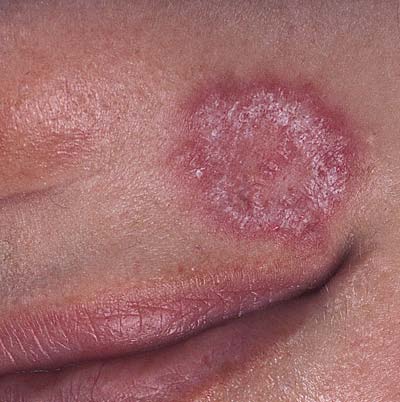
Discoid lesion
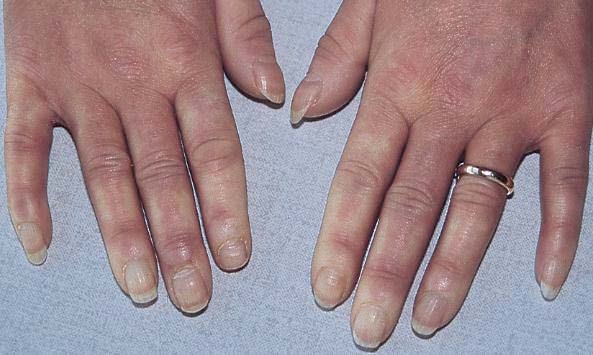
Raynaud phenomenon
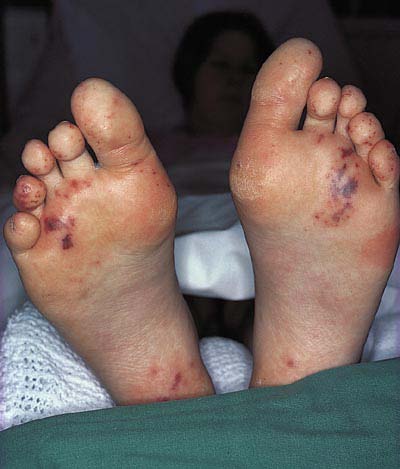
Vasculitis on the feet
Renal involvement is observed in 60% of patients. SLE may manifest
as a monosystemic glomerulonephritis, but usually other organs are
also involved. Glomerulonephritis is an IC-mediated inflammation
with various histological and clinical signs: from mild proteinuria to
rapidly progressive glomerulonephritis. Nephrosis syndrome is very
common.
According to histology, nephritis can be classified. WHO
classification
clinical
manifestations)
I. normal (no alterations)
II. mesangial (mild proteinuria)
III. focal segmental (marked proteinuria, positive sediment)
IV. diffuse (acute nephritis or nephrosis)
V. membranous (nephrosis)
VI. sclerosis/end stage.
New classification of lupus nephritis (ISN/RPS, 2004)
Minimal mesangial lupus nephritis (LN)
Mesangial proliferative LN
Focal LN: III (A)
Active lesions: focal proliferative LN
III (A/C) Active lesions: focal proliferative/sclerosing LN III (C)
Chronic inactive lesions with glomerular scars: focal sclerosing LN
Diffuse LN (diffuse segmental or global) IV-S (A) Active lesions: diffuse segmental proliferative LN IV-G (A) Active lesions: diffuse global proliferative LN IV-S (A/C)
Active and chronic lesions: diffuse segmental
proliferative and sclerosing LN IV-G (A/C)
Active and chronic lesions: diffuse global proliferative
and sclerosing LN IV-S (C) Chronic inactive lesions with scars: diffuse segmental sclerosing LN IV-G (C) Chronic inactive lesions with scars: diffuse global sclerosing LN
Advanced sclerosing LN
Hematological disorders are very common. A mild anemia (anemia of
chronic disease) is always almost present in active disease, frank
hemolysis is rare. Leucopenia is common, but even patients with leukocyte
count <2.0 have no problems at all. Lymphopenia can be profound (0.5),
granulocytopenia rarely results in infections. Thrombocytopenia is more
common in patients with APS.
Central nervous system involvement is common, in particular in patients
with APS. One of the most reliable method to detect CNS involvement is
MRI (however, some foci, seen in MRI may be asymptomatic, therefore the correlation between MRI changes and clinical picture is weak). Psychosis due to corticosteroid treatment is much less frequent that caused by the disease itself. In cuch cases, when in doubt, a bolus corticosteroid is more
likely of benefit then withdawal. There are also minor signs: headache, cognitive and emotional disturbances, e.d. depression, etc.
ACR criteria of CNS involvement:
acute inflammatory demyelinizing polyradiculopathy
(Guillain-Barré syndrome)
aseptic meningitis autonomous nervous system alterations (e.g. orthostatic hypotension) cerebrovascular disease (e.g. stroke) demyelinization syndrome lupus headache mononeuropathy myasthenia gravis cranial neuropathy plexopathy convulsions acute confusion state anxiety cognitive dysfunction mood disorders
Diagnostic criteria of SLE (ARA, 1982, modified in 1997)
1.
Malar rash (or: vespertilio, butterfly rash)
Discoid rash
Oral ulcers: oral or nasopharyngeal ulceration
Arthritis: nonerosive athritis
Serositis: (at least one of the following)
a) pleuritis
b) pericarditis
7. Renal disorder (at least one of the following):
a) persistent proteinuria (>0.5g/day or 3+)
b) cellular casts (erythrocyte, hemoglobin, granular, tubular or mixed)
8. Neurological disorder (at least one of the following):
a) seizures
b) psychosis
9. Hematologic disorder (at least one of the following):
a) hemolytic anemia (with reticulocytosis), or
b) leukopenia (<4.0 on 2 or more occasions), or
c) lymphopenia (<1.5 on 2 or more occasions), or
d) thrombocytopenia (<100 in the absence of offending drug)
10. Immunologic disorder (at least one of the following):
a) abnormal titer anti-dsDNA antibody, or
b) antibody to Sm nuclear antigen, or
c) abnormal titer of anticardiolipin antibody
11. ANA positivity
If 4 criteria are present serially or simultaneously, the diagnosis = SLE
Autoantibodies in SLE
Antigen
Frequency (%)
Native DNA
(double-stranded DNA)
Denatured DNA
(single-stranded DNA)
H1, H2A, H2B, H3, H4)
RNA-protein complexes
nuclear-RNP
(U1-nRNP)
SS-A (Ro)
RNA-protein complexes
SS-B (La)
RNA-protein complexes
* >90% in drug-induced SLE
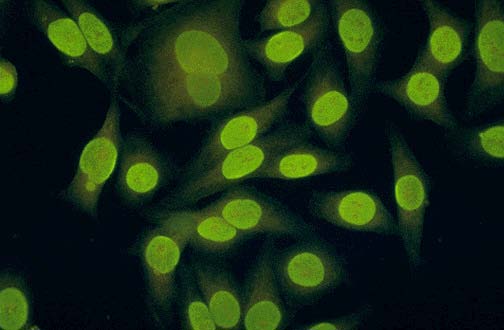
Homogenous ANA positivity on HEp-2 cells
The activity of the disease (monitoring) can be assessed by the
followings:
1. Clinical signs and symptoms are of utmost importance.
2. Blood count: leukopenia cannot be used, but thrombocyto-
penia and hemolysis are of importance.
3. ESR may reflect disease activity, high value usually indicates acivity,
but it remains high in many cases in remission.
4. Renal functions (serum creatinine) and proteinuria should be checked
regularly. High creatinine and an increase in daily proteinuria
usually indicate an activation.
5. Anti-DNA level runs parallel with activity, but it may remain elevated
in remission.
6. Complement activity and C3, C4 levels also indicate activity, they
decrease during activity, and tend to normalize in remission.
7. CRP usually remains normal, an increase may indicate bacterial
infection.
8. There are indices to measure overall disease activity, e.g. SLEDAI
(SLE disease activity index).
Therapy of SLE
1) Inactive
Check-up: 3 months: blood count, urine, creatinine,
blood pressure, once a year: immunology (anti-DNA
complement, anti-cardiolipin etc.
2) moderately active (complaints, laboratory abnormalities,
no general signs)
Organ involvement:
a) skin and/or joints and/or moderate serositis
NSAID and/or (hydroxy)chloroquine
RESPONSE continue chloroquine for at least 6 mo
(fundus control after 2 months, then every 6 months)
NSAID as required
NO RESPONSE 30 mg prednisolone/day (24 mg methyl-
prednisolone) for 1 week, then slowly (during 2 weeks)
taper to 5-10 mg maintenance dose;
discontinue if possible.
OR: 7.5 mg methotrexate (MTX) per week (up to 15
mg/week)
b) hematology:
leucopenia: no treatment
anemia: only hemolysis should be treated if HTC<0.3
hemolysis: 30 mg or more prednisolone/day, maintenance
dose, and/or MTX added
thrombocytopenia: if >50: no treatment
if <20 prednisolone 30 mg or more/day, then
maintenance dose
if instable: danazole, vincristine
c) kidney:
normal creatinine, moderate (0.2-1 g/day) proteinuria, minimal
sediment; if stable only observation, biopsy = treatment
options according to WHO grade.
d) ACL positivity without clinical APS: only observation
3) Active (according to both clinical, and laboratory tests)
a) skin and/or arthritis and/or serositis: 30 mg
prednisolon/day (see above)
NO RESPONSE: 64 mg methylprednisolone/day, maintenance
RESPONSE BUT HIGH MAINTENANCE DOSE: 100 mg Imuran or
MTX added
b) progressive nephritis, either diffuse and/or proliferative
histology: 250 mg methylprednisolone/day cca. 1 g
cumulative dose, then tapering to maintenance dose, OR
6 x 1 g bolus steroid slowly tapered to maintenance
plus 600 mg cyclophosphamide (CTX) infusion once a
month for 1-2 years
(or 100 mg orally for 1-2 years – not used any more!)
If CTX cannot be given or proteinuria is refractory:
4 mg/kg/day cyclosporine A for 3-6 mo, then 2 mg/kg/day
maintenance dose (corticosteroid therapy continued).
Oral mycophenolate may substitute for CTX.
c) APS with thrombosis:
2 mg/kg/day prednisolone + LMW heparin, then
maintenence dose + warfarin,
In arterial thrombosis: aspirin
in severe cases: high dose IVIG
d) severe hemolysis, or CNS involvement:
high dose (250 mg-1 g/day) corticosteroid
alternative: IVIG, apheresis
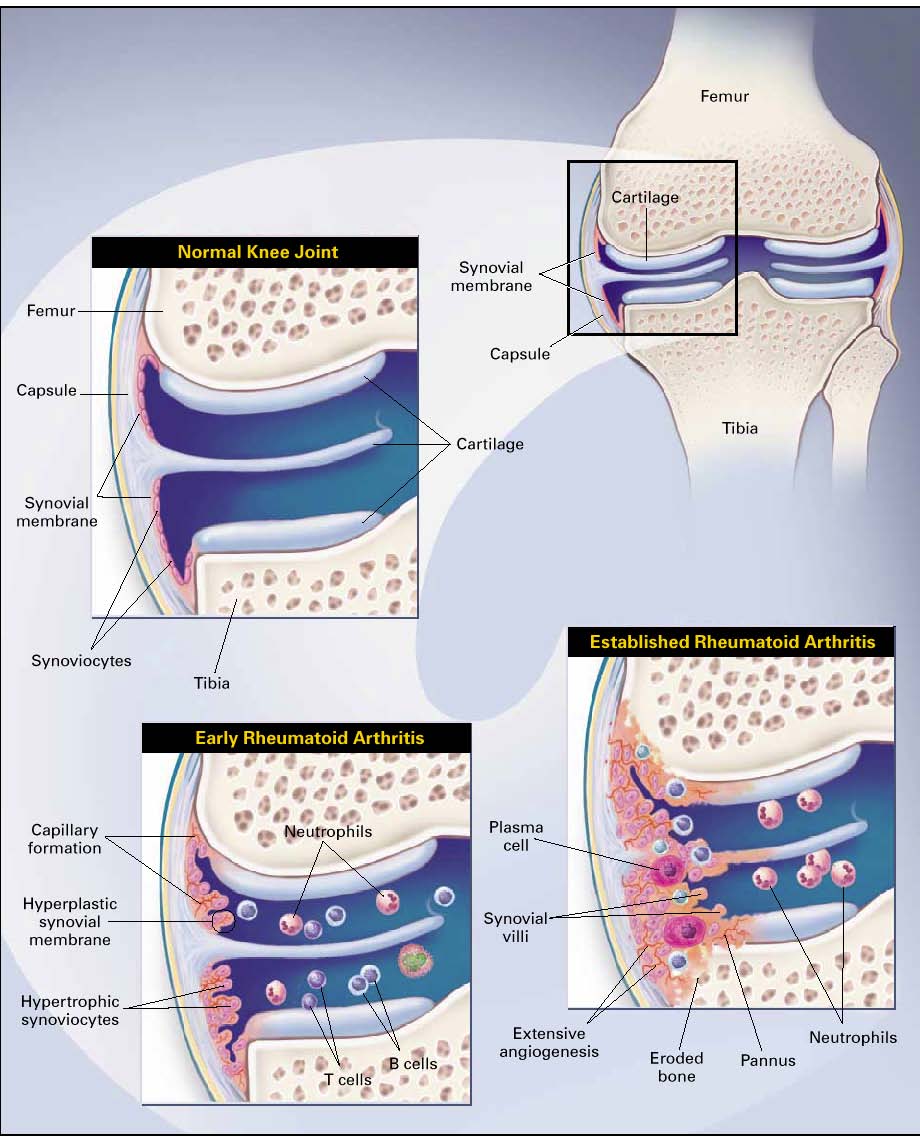
RHEUMATOID ARTHRITIS (RA)
Definition: Chronic destructive diseases characterized
by joint inflammation with pain and swelling. In a
considerable proportion of patients, the arthritis is
progressive, resulting in joint destruction and
ultimately incapacitation and increased mortality.
Epidemiology: relatively common, prevalence: 0.3-1.5
%, the male:female ratio cca. 1:3.
Typical case: woman aged 30-40 years with polyarthritis
and early joint deformities.
Classification Criteria RA (ACR/EULAR, 2010)
Classifiable diseases:
•
inflammation/swelling (synovitis) at least in one joint
other causes of inflammation can be excluded
Criteria (scores to be added) :
A. Joint involvement:
1 large joint
2-10 large joints
1-3 small joint(s) (with/without large joints)
4-10 small joints (with/without large joints)
>10 joints (at least 1 small joint)
B. Serology (at least 1 required)
negative RF or ACPA
low positive RF or ACPA
high positive RF or ACPA
C. Acute phase protein (at least 1 required):
normal CRP or ESR
abnormal CRP or ESR
D. Symptom duration:
<6 weeks
≥6 weeks
At least 6/10 scores are required for a diagnosis
Joint involvement in RA
The most specific sign of RA is arthritis.
It is progressive and deforming in the majority
(2/3) of cases (= erosive polyarthritis)
RA early stage
Early assymmetric RA
PIP joint involvement in RA
RA: swan neck deformity
RA: ulnar deviation
RA: Boutonnière deformity
Involvement of joints of feet in RA
RA – end stage
Periarticular osteoporosis (decalcification)
Erosions and sclerosis (in late stage)
Erosion in RA
Baker's cyst
Bursitis in the shoulder
Bursitis and rheumatoid nodule
Atlantoaxial subluxation
Extraarticular manifestations of RA
• rheumatoid nodules
– subcutaneous
- in internal organs (lung,
aortic valve)
• pleuritis/pericarditis
• fibrotizing alveolitis
• Felty's syndrome
• vasculitis
• amyloidosis
Rheumatoid nodules
Systemic
manifestations of
RA:
pulmonary fibrosis
Systemic
manifestations of
RA:
Caplan's syndrome
Rheumatoid nodules in the lungs
Vasculitis in RA
Disease modifying antirheumatic drugs (DMARDs):
Drug
Adverse effects
gold (i.m.)
dermatitis, stomatitis,
25-50 mg /2-4
proteinuria, enterocolitis,
gold (p.o.)
not used, because of lower tolerability
chloroquine (hydroxy- retinopathia, pigment-
250 mg/day
anomalies
Regular ophthalmology check is required
azathioprine
hepatitis, bone marrow depression
Scarcely given in RA
methotrexate
hepatotoxicity, pulmonary fibrosis,
bone marrow depression
most frequently used therapy
nausea, vomiting
1,5-2 g/day
diarrhea, bone marrow depression
cyclosporine A
nephrotoxicity, tremor
1,5-4 mg/kg/day
creatinine and blood pressure should be
checked regularly
leflunomide
hepatotoxicity, GI
10-20 mg/day
complaints
TNF-α blockers:
local reaction, autoimmune disease (SLE, SM)
infection (tbc)
infliximab, adalimumab
golimumab, certolizumab
pegol
etanercept: 25 mg 2x weekly s.c.
infliximab: 3 mg/kg every 8 week i.v.
adalimumab: 1/week
golimumab: 1/2 weeks
Other:
anakinra (IL-1β blocker)
rituximab (anti-CD20 antibody)
abatacept (T cell activation blocker antibody)
tocilizumab (anti-IL-6R)
Diseases related to RA:
Juvenile forms (= juvenile idiopathic arthritis (JIA)
Subgroups:
a) systemic (Still's disease)
b) pauciarticular (<4 joints)
c) polyarticular (similar to adult RA)
Classification criteria of JIA (ARA, 1982)
1. Persistent arthritis of at least 6 weeks duration in one or more
2. Exclusion of other causes of arthritis (in particular): a. other systemic diseases (SLE, rheumatic fever, vasculitis,
PSS, SS, MCTD, Behçet's syndrome, PM/DM, SPA, Reiter's syndroma, psoriatic arthritis)
b. Infectious arthritis c. Inflammatory bowel diseases d. Neoplasms (e.g. leukemia) e. Nonrheumatic conditions f. Hematologic diseases g. Psychogenic arthralgia h. Other (sarcoidosis, hyperthrophic osteoarthropathy,
villonodular synovitis, chronic active hepatitis, familial Mediterranean fever)
Child with advanced polyarticular JIA
Micrognathia in JIA
Typical skin rash in Still's disease
Exanthema in the rare adult onset Still's disease
Scleroderma
Definition: Inflammatory/degenerative disorder of the connective tissue
with concomitant fibrsis (sclerosis). Skin, vessels and muscles are
involved primarily, often with visceral (GI, kidney) involvation.
Classification:
1. Diffuse cutaneous scleroderma (= progressive systemic sclerosis
2. Limited cutaneous scleroderma (acrosclerotic forms)
3. Overlap syndromes (mixed connective tissue disease,
undifferentiated connective tissue disease)
4. Localised scleroderma (morphea and linear scleroderma)
Epidemiology: incidence 19/1 million, prevalence 19-75/100,000. More
frequent in females; in the 30-55 y age group female/male ratio: 7-
12:1.
Raynaud's phenomenon
Dilated capillary loops in scleroderma
Morphea, with inflamed skin around the lesion
Morphea – late (cicatrizing) stage
Morphea – „lilac ring"
Linear scleroderma
Acrosclerosis – severe form
Ischemic necrosis in acrosclerosis
Atrophic scars in acrosclerosis
Calcinosis
in scleroderma
Telangiectasias in scleroderma
Typical face in systemic scleroderma
Centromere antibody in acrosclerosis
Nucleolar antibody in scleroderma
Bibasilar pulmonary
fibrosis in systemic
scleroderma
Classification criteria of scleroderma (ARA, 1980)
A/ Major criterium:
1. Proximal scleroderma: symmetric thickening, tightening, and induration of the skin
of the fingers and the skin proximal to the MCP or MTP joints. The changes may
affect the entire extremity, face, neck, and trunk (thorax and abdomen).
B/ Minor criteria:
2. Sclerodactyly: as above limited to the fingers.
3. Digital pitted scars or loss of substance from the fingerpad: depressed areas at tips
of fingers or loss of digital pad tissue as a result of ischemia.
4 Bibasilar pulmonary fibrosis: bilateral reticular pattern of linear or lineonodular
densities most pronounced in basilar portions of the lungs on standard chest
roentgenogram: may assume appearance of diffuse mottling or "honeycomb" lung.
These changes should not be attributable to primary lung disease.
Definite diagnosis requires the major and 2 minor criteria.
SCLERODERMA-LIKE DISEASES
EOSINOPHIL FASCIITIS: (Shulman's syndrome)
diffuse fasciitis with eosinophilia
MIXED CONNECTIVE TISSUE DISEASE (MCTD ) (Sharp's syndrome):
A mixture of symptoms of SLE, scleroderma, PM, RA (SS)
The most frequent and most specific signs are as follows:
Raynaud's phenomenon
synovitis: arthritis/arthralgia
sausage-like swollen fingers and hands, and/or sclerodactyly
esophageal dysmotility (dysphagia)
myositis (elevated CPK)
pneumonitis, pulmonary fibrosis.
In addition, depending on the ovelapping disease, serositis, hematologic
signs, etc. may be present.
"OVERLAP" SYNDROMES AND UNDIFFERENTIATED CONNECTIVE
DISEASE (UCTD) = oligosymptomic SLE/RA and scleroderma,
diseases in evolution
Therapy of scleroderma
1) vasodilators (Ca-channel blockers, pentoxyfillin,
2) GI tract: reflux - metoclopramide, proton pump-inhibitors;
blind loop syndrome – octreotide, antibiotics
3) pulmonary hypertension: bosentan, phosphodiesterase
inhibitors ( e.g. sildenafail, tadalafin)
prostacyclin infusion
4) pneumonitis/fibrosis: corticosteroid/cytostatics
5) kidney: ACE-inhibitors
(Idiopathic Inflammatory Myopathies)
Rare disease (prevalence: 5-10/1 million) with a
male:female ratio of 2:1, characterized by proximal
symmetrical muscle weakness (in DM also with heliotrope
rash).
Classification:
I. Adult polymyositis (PM)
II. Adult dermatomyositis (DM)
III. Amyopathic dermatomyositis
IV. Childhood myositis
V. Myositis associated with malignancy
VI. Myositis associated with other systemic autoimmune
disease (e.g. scleroderma, SLE)
VII. Inclusion body myositis.
DM – exanthema on the elbow
Gottron's sign in DM
Gottron's papule
Splinter hemorrhage in myositis
Periorbital exanthema
(heliotrope rash) in
DM
Diagnostic criteria of PM/DM (Bohan & Peter, 1975)
1. symmetrical proximal muscular weakness
2. elevated serum enzymes (CPK, LDH, transaminases,
aldolase)
3. Characteristic triad by EMG:
a) small amplitude, short polyphasic waves,
b) fibrillation, irritability,
c) spontaneous, bizarre discharges
4. Biopsy (=infiltration, necrosis, degenerative-
regenerative signs
5. Heliotrope rash*
--------
* Gottron papules, or Gottron signs are considered more
specific.
Myositis-specific autoantibodies
a) anti-aminoacyl-tRNA synthetase antibodies:
anti-histidil- (= Jo-1)
(= PL-12)
anti-threonil- (= Pl-7)
anti-isoleucil-(= OJ)
b) anti- SRP
(‘signal recognition particle'),
c) other antibodies:
anti-Mi-2 , anti-MAS
Therapy
1) early diagnosis – early therapy!
2) high-dose corticosteroid (CS)
3) in DM, IVIG
4) Imuran or methotrexate
5) Other: cyclosporin, biological therapy
Source: http://www.bel1.sote.hu/upload/seaok1bel/document/Autoimmunity_2013_Gergely_Peter.pdf
Nº 28.430 - marzo 12 de 2012 otorgamiento de preferencias fijas durante dicha etapa facilitará posteriores negociaciones para la creación de un Área de Libre MINISTERIO DE RELACIONES EXTERIORES Que se han realizado las negociaciones necesarias para implementar el otorgamiento de preferencias y establecer disciplinas comerciales entre las Partes; Ratifícase el Acuerdo Preferencial de Comercio entre el MERCOSUR y la
Making music in West London ST MATTHEW'S CONCERT CHOIR AND ORCHESTRA Soloists from the Guildhall School of Music and Drama Stabat Mater Opera choruses and Arias SUNDAY 3RD APRIL 7.30PM St Matthew's Church North Common Road Ealing W5 2QA St Matthews Concert Choir


