
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Pone.0019103 1.1
Amplification of the Angiogenic Signal through theActivation of the TSC/mTOR/HIF Axis by the KSHV vGPCRin Kaposi's Sarcoma
Bruno C. Jham1, Tao Ma1, Jiadi Hu1, Risa Chaisuparat1, Eitan R. Friedman1, Pier Paolo Pandolfi4,
Abraham Schneider1,3, Akrit Sodhi5, Silvia Montaner1,2,3*
1 Department of Oncology and Diagnostic Sciences, School of Dentistry, University of Maryland, Baltimore, Maryland, United States of America, 2 Department of
Pathology, School of Medicine, University of Maryland, Baltimore, Maryland, United States of America, 3 Greenebaum Cancer Center, University of Maryland, Baltimore,
Maryland, United States of America, 4 Cancer Genetics Program, Beth Israel Deaconess Cancer Center, Department of Medicine, Beth Israel Deaconess Medical Center,
Harvard Medical School, Boston, Massachusetts, United States of America, 5 Wilmer Eye Institute, Johns Hopkins School of Medicine, Johns Hopkins University, Baltimore,
Maryland, United States of America
Background: Kaposi's sarcoma (KS) is a vascular neoplasm characterized by the dysregulated expression of angiogenic andinflammatory cytokines. The driving force of the KS lesion, the KSHV-infected spindle cell, secretes elevated levels of vascularendothelial growth factor (VEGF), essential for KS development. However, the origin of VEGF in this tumor remains unclear.
Methodology/Principal Findings: Here we report that the KSHV G protein-coupled receptor (vGPCR) upregulates VEGF inKS through an intricate paracrine mechanism. The cytokines secreted by the few vGPCR-expressing tumor cells activate inneighboring cells multiple pathways (including AKT, ERK, p38 and IKKb) that, in turn, converge on TSC1/2, promoting mTORactivation, HIF upregulation, and VEGF secretion. Conditioned media from vGPCR-expressing cells lead to an mTOR-dependent increase in HIF-1a and HIF-2a protein levels and VEGF upregulation. In a mouse allograft model for KS, specificinhibition of the paracrine activation of mTOR in non-vGPCR-expressing cells was sufficient to inhibit HIF upregulation inthese cells, and abolished the ability of the vGPCR-expressing cells to promote tumor formation in vivo. Similarly,pharmacologic inhibition of HIF in this model blocked VEGF secretion and also lead to tumor regression.
Conclusions/Significance: Our findings provide a compelling explanation for how the few tumor cells expressing vGPCRcan contribute to the dramatic amplification of VEGF secretion in KS, and further provide a molecular mechanism for howcytokine dysregulation in KS fuels angiogenesis and tumor development. These data further suggest that activation of HIFby vGPCR may be a vulnerable target for the treatment of patients with KS.
Citation: Jham BC, Ma T, Hu J, Chaisuparat R, Friedman ER, et al. (2011) Amplification of the Angiogenic Signal through the Activation of the TSC/mTOR/HIF Axisby the KSHV vGPCR in Kaposi's Sarcoma. PLoS ONE 6(4): e19103. doi:10.1371/journal.pone.0019103
Editor: Lin Zhang, University of Pennsylvania, United States of America
Received February 25, 2011; Accepted March 16, 2011; Published April 29, 2011
Copyright: ß 2011 Jham et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was supported by grant R01CA119911 (National Cancer Institute, National Institutes of Health). The funders had no role in study design, datacollection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist.
* E-mail:
[email protected]
VEGF is expressed at elevated levels by KSHV-infectedendothelial cells in vitro, and by KS spindle cells in vivo; a strict
Kaposi's sarcoma (KS) is a multifocal vascular neoplasm
requirement for VEGF has also been demonstrated for KS spindle
invariably associated with infection with the KS-associated human
cells grown in vitro [3,4,5,6]. These observations are suggestive of
herpesvirus (KSHV/HHV8), which is characterized by cytokine
an autocrine mechanism for this angiogenic factor in the growth of
dysregulation [1]. The driving force of the KS lesion is the KSHV-
this vascular tumor. Indeed, like VEGF, its cognate receptor,
infected spindle-shaped tumor (spindle) cell, thought to have a
VEGFR2 (KDR), is also upregulated in AIDS-KS primary lesions
vascular endothelial or endothelial precursor origin. The promo-
as well as in AIDS-KS spindle cell cultures [4,7,8]. However,
tion of the angiogenic phenotype in these lesions is supposed to be
reasonable disagreement remains as to the precise molecular
mediated by elevated levels of pro-inflammatory and pro-
mechanism whereby KSHV promotes VEGF secretion.
angiogenic secretions (cytokines, chemokines and growth factors)
Of interest, KSHV ORF74 encodes for a viral G protein-coupled
from the KS spindle cells [1]. Indeed, KS is often thought to result
receptor (vGPCR) with close homology to the mammalian CXCR1
from reactive endothelial hyperproliferation induced by the
and CXCR2 and ligand-independent (constitutive) activity [9,10].
chronic release of these molecules and has served as a model for
When expressed upon endothelial-specific retroviral infection or as a
tumor- and inflammation-induced angiogenesis [1,2].
transgene, vGPCR is sufficient to recapitulate KS-like lesions in mice
Among the angiogenic factors elaborated by the KS spindle
[11,12,13]. Indeed, emerging data suggest that vGPCR may play a
cells, VEGF is unique for its profound impact on KS pathogenesis.
role in KS initiation, progression and maintenance [12,14,15].
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
vGPCR has proven to be a powerful oncogene and a potent
vivo (data not shown; [15]). However, these cells (EC-vCYC/
angiogenic activator [1]. Expression of vGPCR in endothelial cells
vFLIP) are able to induce allografts in nude mice when co-injected
activates intracellular pathways that induce cell survival and
with vGPCR-expressing cells (EC-vGPCR) (Fig. 1E). Immunohis-
transformation. In addition, vGPCR-expressing cells elaborate
tochemical staining of these lesions also shows upregulation of
pro-angiogenic factors that are thought to promote the recruit-
VEGF in most tumor cells (Fig. 1E). Interestingly, when these
ment and transformation of neighboring endothelial cells [16].
mixed-cell allografts are treated with Rapamycin, which is able to
Both VEGF and KDR have been previously reported to be
block vGPCR tumorigenesis [19], a reduction in VEGF expression
upregulated by cells expressing vGPCR [7,8,17,18]. However, the
in treated tumors is observed. Collectively, these results suggest
limited expression of this viral oncogene in only a few tumor cells
that the paracrine upregulation of VEGF by vGPCR requires the
in both transgenic KS mouse models and human KS tissues raises
activation of the mTOR signaling cascade in vitro and in vivo.
the question as to the relative contribution of vGPCR to VEGFsecretion, suggesting that the relationship between vGPCR and
vGPCR paracrine secretions activate mTOR through
VEGF is not clearly established. Here we set out to further
multiple signaling pathways
characterize the contribution of vGPCR to the upregulation of
Diverse extracellular stimuli influence mTOR activity through
VEGF secretion in KS.
posttranslational modification of TSC1/TSC2 [20]. Indeed,numerous kinases are able to phosphorylate TSC1 or TSC2,
integrating extracellular signals and tumorigenesis through thecontrol of TSC1/TSC2/mTOR [21]. Among these kinases,
vGPCR activates VEGF expression through a paracrine
AKT, ERK, and p38/MK2 have been shown to phosphorylate
mechanism dependent on mTOR
TSC2 in specific sites, including Thr1462, Ser664 and Ser1254,
The KSHV-encoded vGPCR causes angioproliferative lesions
respectively [22,23,24,25,26]. Conversely, IKKb induces mTOR
remarkably similar to human KS, when expressed upon
activation by phosphorylating TSC1 in Ser487 and Ser511 [27].
endothelial-specific retroviral infection in immunocompetent
We have previously shown that induction of AKT activity by
animals (Fig. 1A–B) [12]. Interestingly, immunohistochemical
vGPCR promotes mTOR activation in neighboring cells [19].
staining of these vGPCR tumors using a specific vGPCR antibody
However, the contribution of other kinases upstream of TSC/
reveals the presence of this viral protein in only a small percentage
mTOR to vGPCR oncogenesis remains unclear. We thus treated
of tumor cells, similar to the expression pattern of vGPCR in
HMEC1 with media conditioned by control or vGPCR-expressing
human KS, suggestive of a paracrine contribution of vGPCR to
cells and assessed the activation of TSC kinases by these
Kaposi's sarcomagenesis (Fig. 1B) [12,13]. In this regard, vGPCR
supernatants. In addition to AKT, we found that ERK, p38 and
has been implicated in the induction of the expression of VEGF, a
IKKb were also activated by vGPCR paracrine secretions
key angiogenic factor highly upregulated in KS [17,18]. However,
(Fig. 2A). Activation of these kinases correlated with the
staining of vGPCR tumors – and human KS – with a specific
phosphorylation of TSC2 in Thr1462, Ser664 and Ser1254, and
antibody against VEGF reveals a robust expression of this factor in
phosphorylation of TSC1 in Ser511, respectively, and the
most tumor cells (Fig. 1B), indicating that vGPCR may also
upregulation of mTOR activity, assessed by S6K phosphorylation
upregulate VEGF in neighboring cells through an indirect
(Fig. 2A). We also evaluated the activation of these TSC1/2
mechanism. To explore this possibility, we used an inducible
kinases by individual GPCR angiogenic factors [14,28]. Figure S1
(Tet-on) expression system for vGPCR in immortalized human
shows that all the cytokines tested were able to induce
microdermal endothelial cells (HMEC1). Using this system, we
phosphorylation of TSC1 and/or TSC2. Interestingly, IL-1b,
confirmed that induction of vGPCR expression in endothelial cells
IL-10, TNFa and VEGF are each able to promote phosphory-
leads to the potent upregulation of VEGF, as previously reported
lation of TSC1/2 on at least three separate sites (Fig. S1).
(Fig. 1C) [17,18]. Moreover, we observed that exposure of
Collectively, these results suggest that the secreted factors
HMEC1 to media conditioned by these vGPCR-expressing cells
elaborated by vGPCR-expressing cells act together to upregulate
(vGPCR CM) is similarly able to promote VEGF expression
multiple intracellular signaling pathways that converge on TSC/
We therefore set out to determine the mechanism whereby
To determine the relative contribution of these TSC kinases to
vGPCR angiogenic factors can induce VEGF upregulation. In this
the paracrine mTOR activation by vGPCR, we used specific
regard, we have previously reported that vGPCR paracrine
inhibitors of PI3K/AKT, MEK/ERK, p38 or IKKb. Surprising-
secretions activates TSC/mTOR, a signaling route that has been
ly, we observed that either pharmacological inhibition of AKT-
shown to regulate the expression of VEGF [19,20]. We thus
mediated phosphorylation of TSC2 or IKKb-mediated phosphor-
treated HMEC1s with supernatants derived from vGPCR-
ylation of TSC1 lead to a complete inhibition of mTOR,
expressing cells (vGPCR CM) or control cells (Control CM), in
measured by S6K phosphorylation (Fig. 2A). Conversely,
the absence or the presence of the mTOR inhibitor, Rapamycin.
pharmacological inhibition of either ERK or p38 leads to only
Figure 1D shows that exposure of endothelial cells to vGPCR
partial inhibition of ERK-mediated or p38-mediated S6K
secreted factors leads to the upregulation of the transcription and
phosphorylation, respectively. We then used siRNA to specifically
translation of VEGF in a Rapamycin-sensitive manner.
knock-down expression of AKT, ERK1 and 2, p38, or IKKb.
To further evaluate the contribution of the vGPCR-induced
Knock-down of these kinases was only sufficient to partially inhibit
paracrine activation of mTOR to VEGF upregulation in vivo, we
mTOR (Fig. 2B). Collectively, these results suggest that there is a
used an allograft model in which (SV-40) immortalized murine
redundancy in the pathways leading to the phosphorylation of
endothelial cells (SVECs) expressing vGPCR (EC-vGPCR) are
TSC1 and 2 and the activation of mTOR by the paracrine
mixed with SVECs co-expressing two non-tumorigenic KSHV
secretions of vGPCR-expressing cells.
latent genes, vCyclin and vFLIP (EC-vCYC/vFLIP), in a (1:10)
We then stained murine vGPCR tumors and human KS tissues
ratio that approximates the proportion of expressing cells in
with specific antibodies against the phosphorylated (activated)
human KS (Fig. 1E) [15]. Cells expressing vCYC and vFLIP do
forms of AKT, ERK, p38 or IKKb or against the corresponding
not show VEGF upregulation in vitro nor are they tumorigenic in
TSC2/TSC1 phosphorylated form, induced by each kinase
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
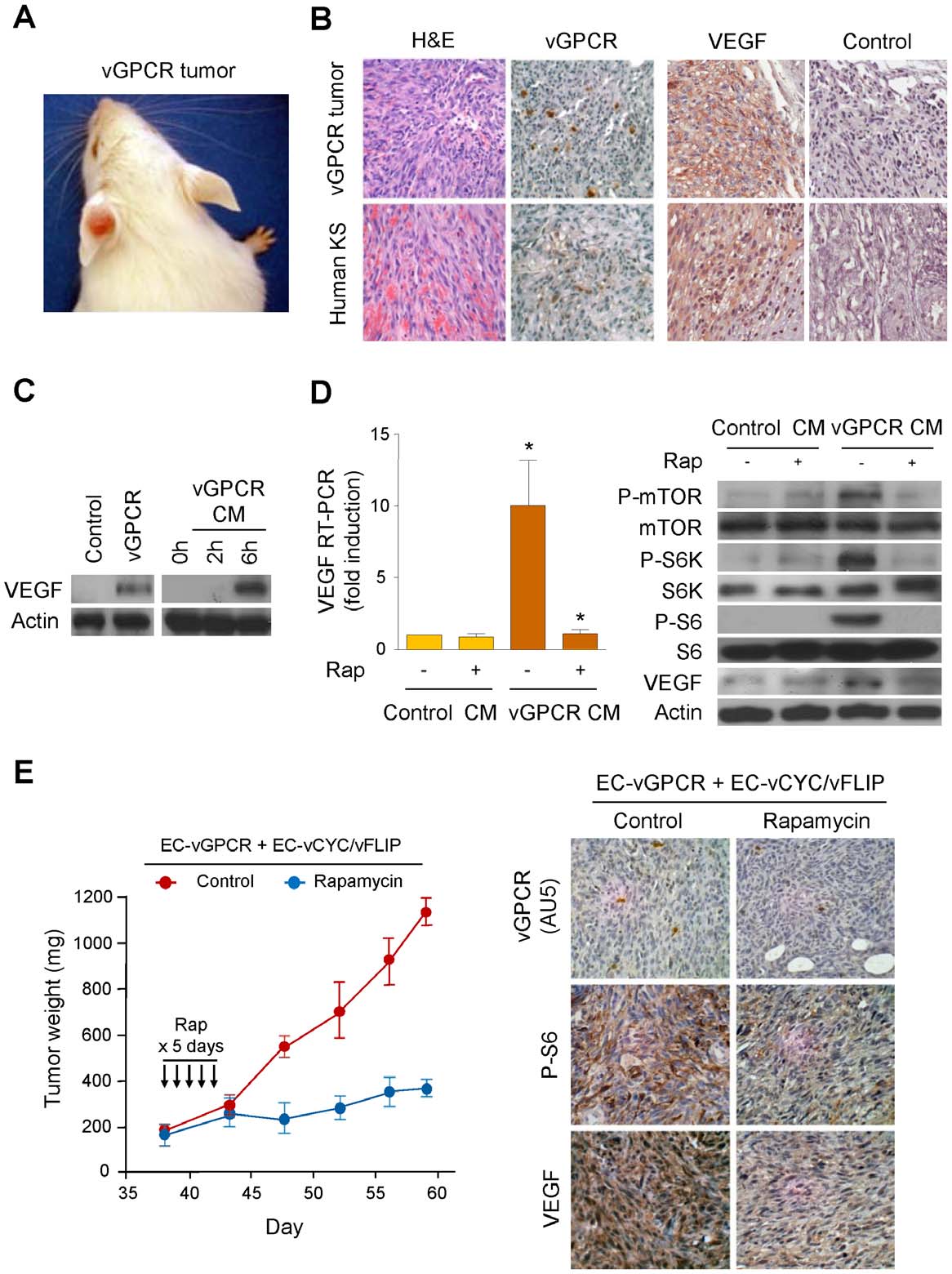
VEGF Amplification by vGPCR Activation of HIF
Figure 1. vGPCR activates VEGF expression through an mTOR-dependent paracrine mechanism. (A) KS-like lesion developed uponretroviral transduction of vGPCR in TIE2-tva mice (vGPCR tumor), as described in Materials and Methods. (B) H&E and immunohistochemical stainingsof vGPCR tumor and human KS, using antibodies against vGPCR, VEGF or an isotype-matched control antibody. (C) Upregulation of VEGF in HMEC1stransfected with Tet REV TA and pBIG AU5 vGPCR and treated with doxycycline (vGPCR), respect to untreated cells (Control). VEGF upregulation inHMEC1 exposed for 2 h or 6 h to supernatants collected from vGPCR-expressing cells (vGPCR CM). (D) Supernatants from vGPCR-expressing cells(vGPCR CM) or control cells (Control CM) were used to treat HMEC1 in the presence or absence of Rapamycin (50 nM). RT-PCR and Western blotanalysis were used to determine levels of VEGF mRNA and protein, respectively. (E) EC-vGPCR (10%) were mixed with EC- vCYC/vFLIP (90%), andinjected into athymic nu/nu mice for allograft formation. Tumor weight curves, and immunohistochemical detection in tumor tissue of (AU5-tagged)vGPCR-expressing cells, phosphorylated ribosomal S6 protein or VEGF, upon treatment with (10 mg/kg) Rapamycin of vehicle (Control), are shown.
doi:10.1371/journal.pone.0019103.g001
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
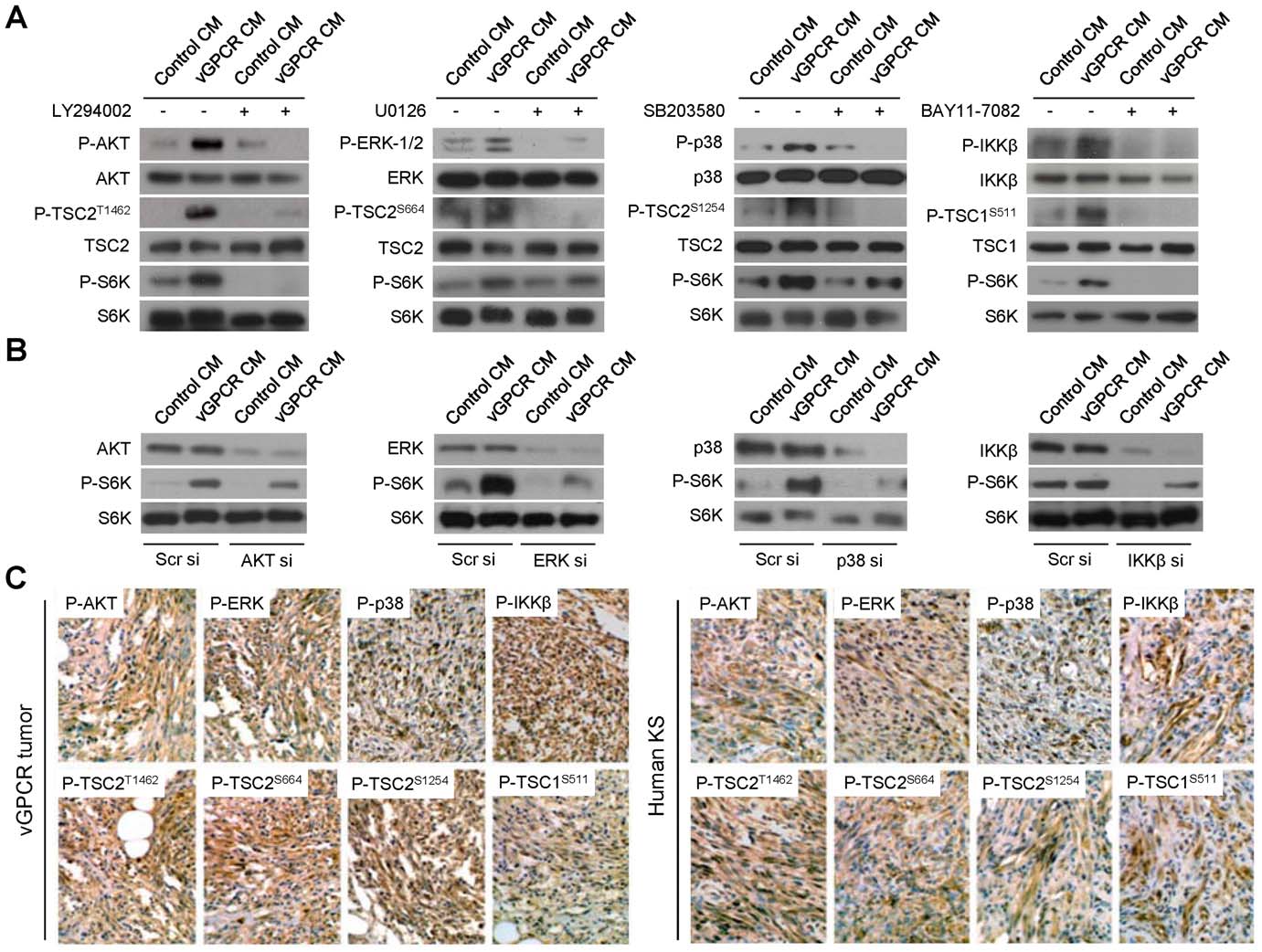
VEGF Amplification by vGPCR Activation of HIF
Figure 2. vGPCR secretions regulate TSC/mTOR through multiple signaling pathways in vitro and in vivo. (A) HMEC1s were pretreatedwith vehicle or inhibitors of the AKT, ERK, p38 or IKKb pathways, LY294002 (50 mM), U0126 (50 mM), SB203580 (50 mM) or BAY11-7082 (40 mM). Cellswere then exposed to media conditioned by control cells (Control CM) or vGPCR-expressing cells (vGPCR CM). Phosphorylation levels of thecorresponding kinase (AKT, ERK1/2, p38 or IKKb), TSC2/1 targeted phosphorylation site (P-TSC2T1462, P-TSC2S664, P-TSC2S1254 or P-TSC1S511), and S6Kare shown. (B) HMEC1s were transfected with Scrambled siRNA or siRNA for AKT, ERK (ERK1 and 2), p38 or IKKb. Cells were then exposed to mediaconditioned by control or vGPCR-expressing cells. Levels of the corresponding kinase (AKT, ERK1/2, p38 or IKKb) and S6K are shown. (C)Immunohistochemical staining of vGPCR tumors and human KS with antibodies against P-AKT, P-ERK, P-p38 or P-IKKb, and the corresponding TSC2/1targeted phosphorylation site, P-TSC2T1462, P-TSC2S664, P-TSC2S1254 or P-TSC1S511.
doi:10.1371/journal.pone.0019103.g002
(Fig. 2C). In support of our in vitro observations, we found
HMEC1 with media conditioned by vGPCR-expressing cells or
phosphorylation of these four kinases and the corresponding
control cells, in the absence or presence of Rapamycin, and
targeted aminoacids in TSC2 or TSC1, in both vGPCR tumors
examined the levels of HIF-1a and HIF-2a mRNA and protein.
and human KS (Fig. 2C). These findings suggest a role for these
Interestingly, we found that vGPCR angiogenic factors induced an
kinases upstream of TSC/mTOR in vGPCR tumorigenesis and
upregulation of HIF-1a mRNA (3-fold) and HIF-2a mRNA (18-
Kaposi's sarcomagenesis in vivo.
fold); both were blocked by the mTOR inhibitor (Fig. 3A).
Upregulation of HIF-1a, HIF-2a and VEGF protein levels by
Paracrine activation of TSC/mTOR by vGPCR angiogenic
vGPCR secretions was also blocked by Rapamycin (Fig. 3A).
factors results in the upregulation of HIF-1a/2a
Furthermore, the increase in VEGF transcription and translation
TSC/mTOR has been shown to regulate Hypoxia Inducible
by vGPCR supernatants was blocked by the expression of a
Factor (HIF), a family of transcription factors containing an
specific siRNA of HIF-1b (Fig. 3B). When we investigated the
inducible a subunit and a constitutive b subunit [20,29]. HIF
levels of HIF-1a and HIF-2a proteins in vGPCR murine tumors
promotes neovascularization and vascular remodeling by control-
and KS biopsy specimens by immunohistochemical analysis, we
ling the expression of key angiogenic proteins, including VEGF
found that both vGPCR murine tumors and human KS showed a
[30]. Since vGPCR angiogenic factors activate TSC/mTOR
remarkable overexpression of these transcription factors, com-
through a variety of intracellular routes, we next investigated
pared to normal skin (results not shown) (Fig. 3C). Collectively,
whether this activation could lead to the upregulation of VEGF
these results suggest that vGPCR may induce paracrine upregula-
through a HIF-dependent mechanism. To this end, we treated
tion of VEGF through an mTOR/HIF-dependent mechanism.
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
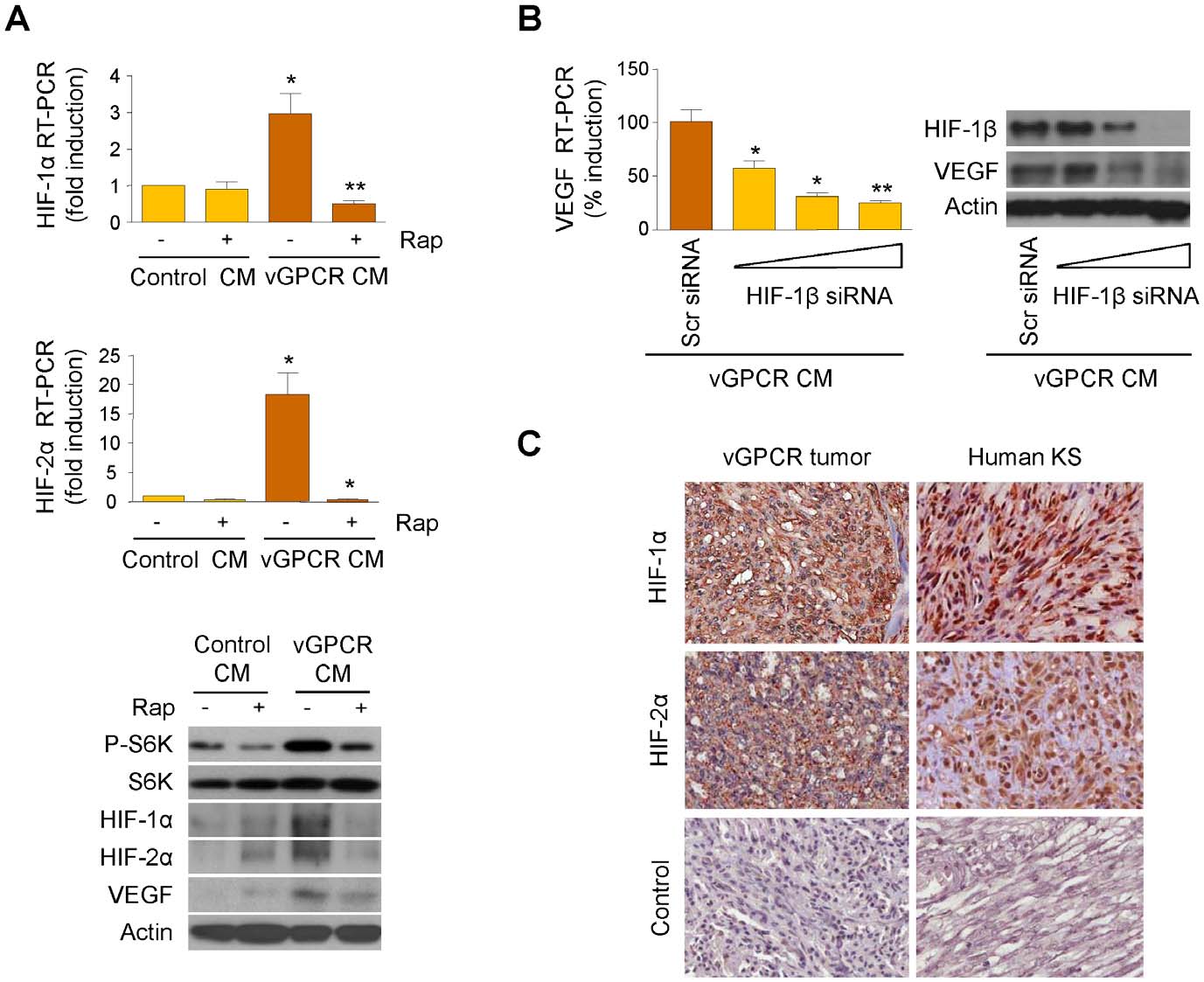
VEGF Amplification by vGPCR Activation of HIF
Figure 3. Upregulation of HIF-1a/2a by vGPCR paracrine secretions. (A) HMEC1s were exposed to media conditioned by control (Control CM)or vGPCR-expressing cells (vGPCR CM), in the presence of vehicle or Rapamycin (50 mM). mRNA levels of HIF-1a and HIF-2a and protein levels of HIF-1a, HIF-2a and VEGF are shown. (B) HMEC1s were transfected with increasing doses (20, 40 and 80 nM) of HIF-1b siRNA or Scrambled siRNA. Cellswere then exposed to media conditioned by vGPCR-expressing cells. VEGF mRNA levels are shown. (C) Immunohistochemical analysis of HIF-1a andHIF-2a, and staining using an isotype-matched control antibody, in vGPCR tumor and human KS.
doi:10.1371/journal.pone.0019103.g003
Paracrine activation of mTOR is required for HIF
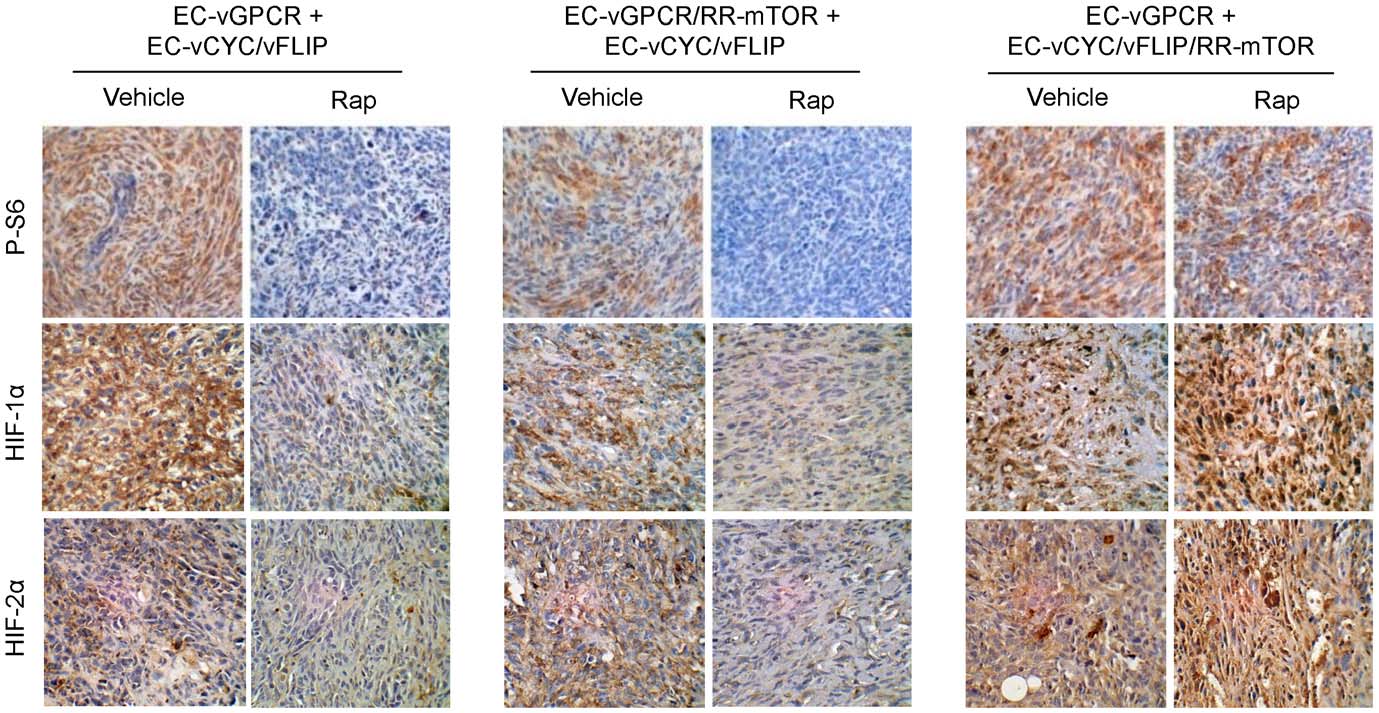
and treated established tumors with Rapamycin or vehicle (Fig. 4).
upregulation in vGPCR sarcomagenesis in vivo
Similar to EC-vGPCR+EC-vCYC/vFLIP tumors, growth of
vGPCR activates mTOR through both direct and indirect
allografts formed upon injection with EC-vGPCR/RR-mTOR +
mechanisms and both may thus contribute to endothelial cell
EC-vCYC/vFLIP was strongly inhibited by treatment with
transformation and angiogenic dysregulation in KS. To assess the
Rapamycin, suggesting that protection from the inhibition of
relative contribution of vGPCR direct versus paracrine activation
direct mTOR activation within vGPCR-expressing cells was not
of TSC/mTOR/HIF to vGPCR oncogenesis, we generated cell
sufficient to render these tumors sensitive to the drug. Conversely,
lines co-expressing vGPCR or vCYC/vFLIP along with a
allografts derived from the injection of EC-vGPCR (10%) + EC-
Rapamycin-Resistant mTOR mutant (RR-mTOR) that bears a
vCYC/vFLIP/RR-mTOR (90%) cells continued growing even
Ser2035RIle (SI) substitution in the FKBP12-Rapamycin-binding
upon treatment with Rapamycin, suggesting that the sensitivity to
domain (EC-vGPCR/RR-mTOR or EC-vCYC/vFLIP/RR-
the drug of these allografts is due to the inhibition of the paracrine
mTOR) [19]. Expression of RR-mTOR strongly protected
activation of mTOR in neighboring cells by the angiogenic factors
vGPCR- and vCYC/vFLIP-expressing cells from the ability of
elaborated by vGPCR-expressing cells (Fig. 4). Of interest, tissue
Rapamycin to inhibit mTOR activation in vitro (data not shown).
staining with a phospho-S6 ribosomal protein specific antibody
We then established mixed-cell allografts injecting athymic nu/nu
confirmed the inhibition of mTOR activity in most cells of EC-
mice with EC-vGPCR (10%) + EC-vCYC/vFLIP (90%) cells, EC-
vGPCR (10%) + EC-vCYC/vFLIP (90%) as well as EC-vGPCR/
vGPCR/RR-mTOR (10%) + EC- vCYC/vFLIP (90%) cells or
RR-mTOR (10%) + EC- vCYC/vFLIP (90%) tumors, but not
EC-vGPCR (10%) + EC- vCYC/vFLIP/RR-mTOR (90%) cells
EC-vGPCR (10%) + EC- vCYC/vFLIP/RR-mTOR (90%)
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
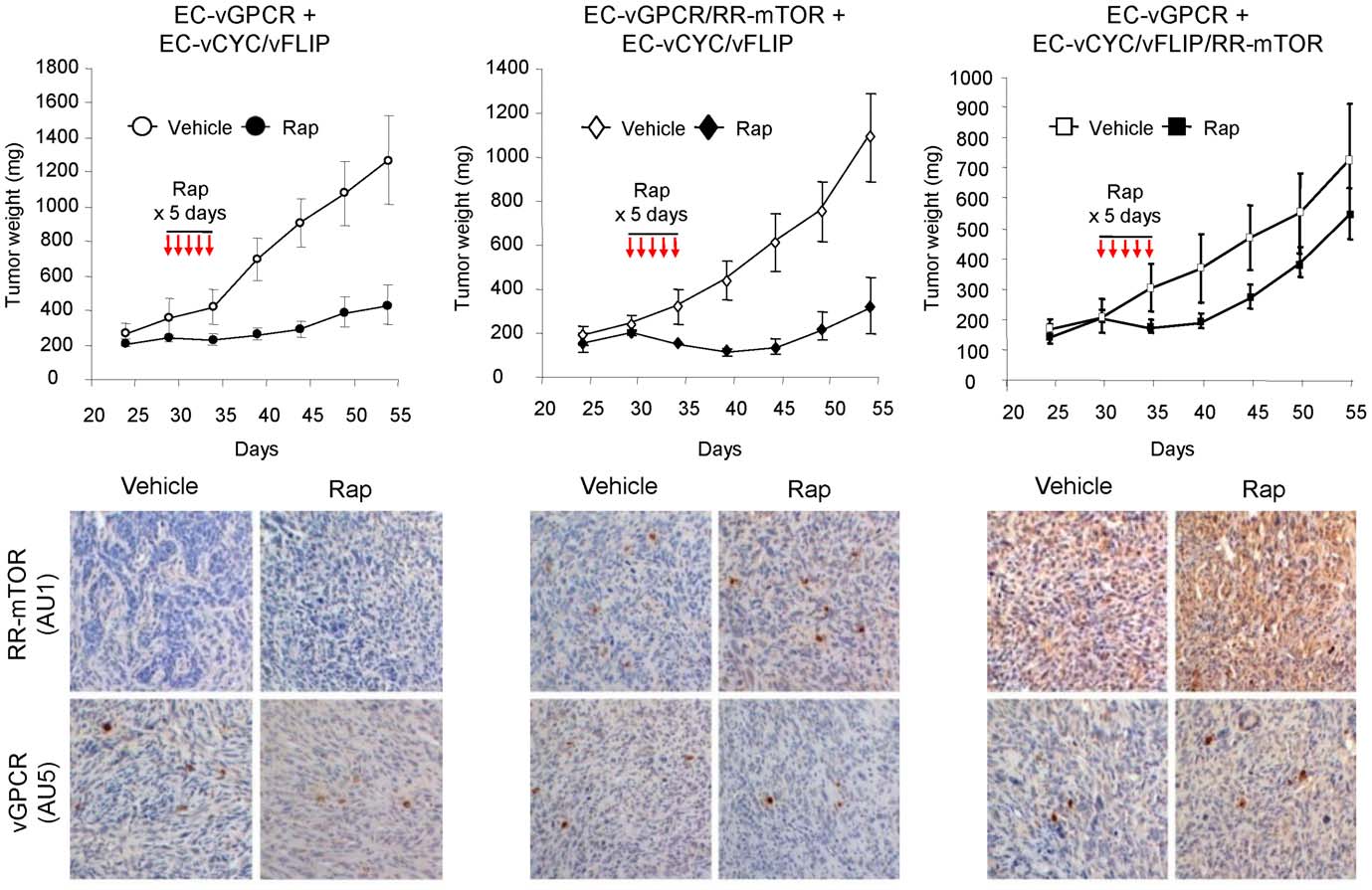
VEGF Amplification by vGPCR Activation of HIF
Figure 4. Paracrine activation of mTOR is required for vGPCR sarcomagenesis in vivo. Tumor allografts were generated upon injection ofathymic nu/nu mice with EC-vGPCR (10%) + EC-vCYC/vFLIP (90%) cells, EC-vGPCR/RR-mTOR (10%) + EC-vCYC/vFLIP (90%) cells or EC-vGPCR (10%) +EC-vCYC/vFLIP/RR-mTOR (90%) cells. Lesions were then treated with (10 mg/kg) Rapamycin or vehicle. Curves of tumor growth andimmunohistochemical staining of tumor tissue with anti-AU1 or anti-AU5 antibodies, revealing expression of RR-mTOR or vGPCR, respectively, areshown.
doi:10.1371/journal.pone.0019103.g004
allografts, upon Rapamycin treatment (Fig. 5). Similarly, HIF
average estimated weight of vehicle-treated tumors was 702 mg (a
levels were reduced in treated tumors but remained elevated in
4.8 fold increase) vs. an average estimated weight of 234 mg (a
most tumor cells of EC-vGPCR (10%) + EC- vCYC/vFLIP/RR-
1.7 fold increase) of Digoxin-treated tumors (Fig. 6). Immuno-
mTOR (90%) allografts even after treatment with Rapamycin
histochemical analysis of these lesions demonstrated a dramatic
(Fig. 5). Collectively, these findings support an essential role of
reduction in the levels of HIF as well as VEGF in the Digoxin-
vGPCR paracrine secretions in the mTOR-driven promotion of
treated animals compared to control mice (Fig. 6). Taken
HIF stabilization and VEGF secretion, and in vGPCR tumori-
together, our data provide the basis for the early assessment of
HIF inhibitors as an anti-KS therapy.
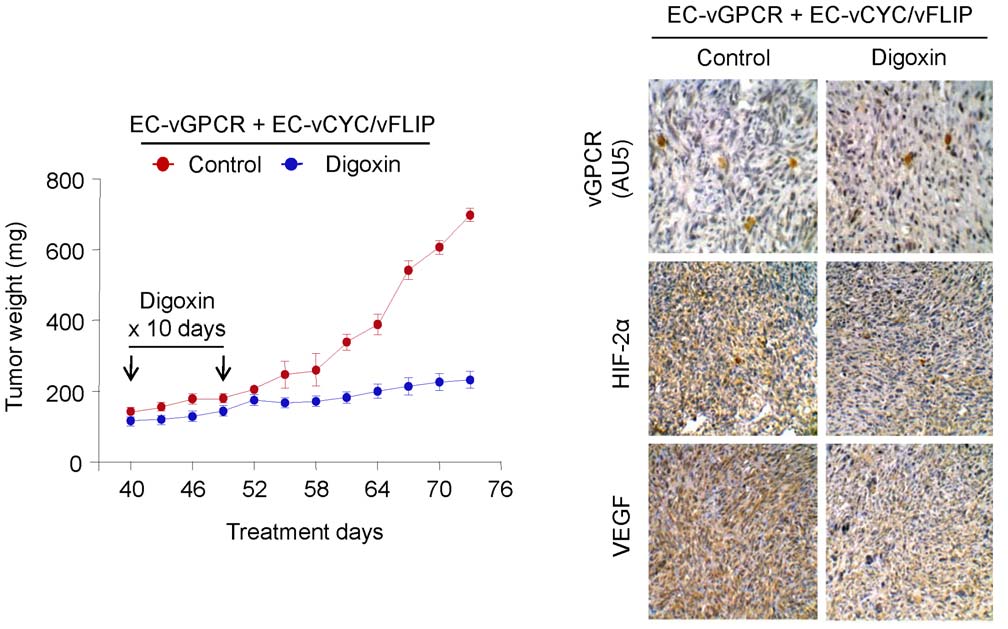
Inhibition of HIF blocks vGPCR tumorigenesis in vivo
Of interest, several drugs have been recently described that
target HIF expression or activity; these drugs have demonstrated
KS is an angioproliferative tumor characterized by the presence
anti-angiogenic and anti-cancer effects in vivo [30]. The
of angiogenic and inflammatory mediators [1]. The observation
identification of HIF as a key factor in vGPCR angiogenic
that KS tumors tend to localize to sites of inflammation suggests
amplification prompted us to explore inhibition of HIF as a
that these lesions thrive in a cytokine-rich environment [1].
potential therapeutic approach for KS treatment. We therefore
Indeed, KS spindle cells do not appear to be truly transformed;
established tumor allografts by injecting mixed-cell populations of
rather, the KS spindle cell elaborates a variety of cytokines,
EC-vGPCR and EC-vCYC/vFLIP cells in athymic nu/nu mice
chemokines and growth factors that are essential for their growth
(Fig. 6). Animals were then treated with either (2 mg/kg)
and survival. Indeed, isolated KS spindle cells remain strictly
Digoxin, a cardiac glycoside that has been shown to inhibit
dependent on cytokines and growth factors to proliferate in vitro
HIF-1a synthesis and block tumor formation, or vehicle (Control)
and are not tumorigenic when tested in animal models. Among the
(Fig. 6) [31]. Drug toxicity, as assessed by weight loss, was
numerous angiogenic mediators on which the KS spindle cell is
minimal in the treated group (reduction ,5%) during the
dependent, VEGF has been shown to be essential for KS spindle
treatment period (results not shown). Inhibition of tumor growth
cell survival in vitro and KS pathogenesis in vivo.
by the treatment with Digoxin was sustained for the duration of
We previously reported a mechanism whereby the KSHV
the experiment. At the end of the study, we observed that the
vGPCR promotes the upregulation of VEGF transcription in
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
Figure 5. Paracrine activation of mTOR is required for HIF upregulation in vGPCR sarcomagenesis. Immunohistochemical staining withspecific antibodies against phospho-S6 ribosomal protein, HIF-1a and HIF-2a of the allografts generated upon injection of EC-vGPCR (10%) + EC-vCYC/vFLIP (90%), EC-vGPCR/RR-mTOR (10%) + EC-vCYC/vFLIP (90%) or EC-vGPCR (10%) + EC-vCYC/vFLIP/RR-mTOR (90%), treated with vehicle or(10 mg/kg) Rapamycin.
doi:10.1371/journal.pone.0019103.g005
vGPCR-expressing cells [17]. We report here that vGPCR also
an explanation for how this unusual viral oncogene can play a role
upregulates VEGF through a complex indirect (paracrine)
in KS despite the observation that its expression is restricted to
mechanism. Upregulation of VEGF in neighboring (non-
only a few tumor cells. vGPCR angiogenic amplification involves
vGPCR-expressing) tumor cells results in a dramatic amplification
the secretion of angiogenic and inflammatory cytokines by
of the angiogenic signal promoted by vGPCR and helps provide
vGPCR-expressing cells which then activate in neighboring cells
Figure 6. Inhibition of HIF blocks vGPCR tumorigenesis in vivo. Tumor allografts were generated upon injection of athymic nu/nu mice withEC-vGPCR (10%) + EC-vCYC/vFLIP (90%) cells. Lesions were treated with (2 mg/kg) Digoxin or vehicle. Tumor growth curves andimmunohistochemical staining of tumor tissue with anti-AU5 (revealing vGPCR-expresing cells), anti-HIF-2a or anti-VEGF antibody, are shown.
doi:10.1371/journal.pone.0019103.g006
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
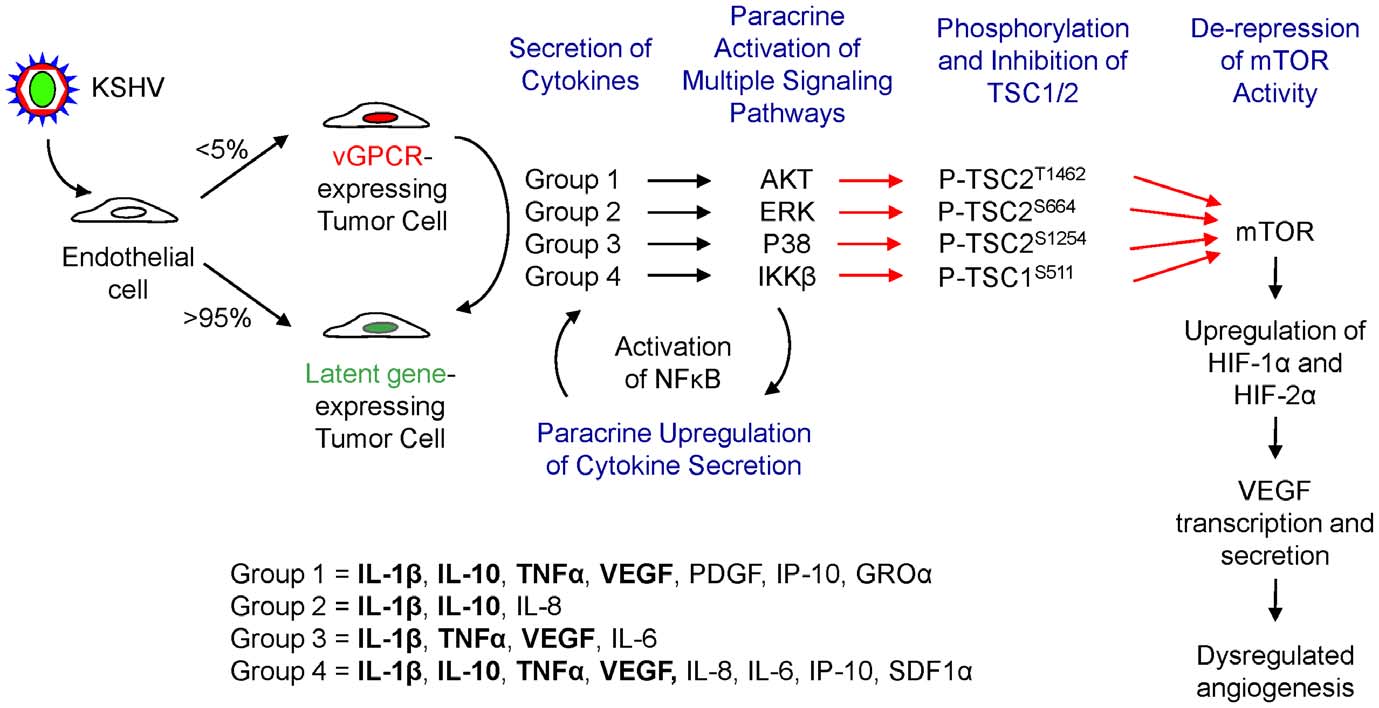
multiple signaling pathways that ultimately converge on TSC1
cytokines that promote the activation of IKKb. Ultimately, further
and 2, resulting in de-repression of mTOR and the promotion of
investigation into the relative contribution of each of these
HIF upregulation and VEGF transcription and secretion (Fig. 7).
cytokines to the angiogenic phenotype in KS as well as in other
TSC kinases activated by vGPCR paracrine secretions include
tumors may be warranted.
AKT, ERK, p38 and IKKb, inducing phosphorylation of TSC2/
Several additional KSHV genes have also been shown to
1 at specific regulatory sites [22,23,24,25,26,27]. Although
upregulate cytokine levels. These viral proteins, including
pharmacological inhibition of AKT-mediated phosphorylation of
vFLIP, kaposin A, kaposin B, K1 and K15, undoubtedly
TSC2 or IKKb-mediated phosphorylation of TSC1 lead to a
contribute to the inflammatory milieu observed in KS
complete inhibition of mTOR activity, siRNA knock down
[33,34,35,36,37,38]. As KSHV infection itself has been
expression of AKT, ERK1 and 2, p38, or IKKb was only
demonstrated to induce cytokine release [1], the relative
sufficient to partially inhibit S6K phosphorylation. This suggests
contribution of each of the KSHV genes to cytokine dysregu-
either that the PI3K/AKT inhibitor, LY294002, and the IKKb
lation and the angiogenic phenotype in KS remains to be
inhibitor, BAY 11-7082, have non-specific inhibitory effects, or
determined. Moreover, various inflammatory cytokines, includ-
that the RNAi knockdown of AKT and IKKb were less efficient
ing IL-1, TNF-a and interferon-c (IFN—c), are increased upon
than their pharmacological inhibition. Although we suspect the
HIV infection, an important cofactor in KS development [39].
former to be true, regulation of mTOR through these pathways
Collectively, these findings suggest that cytokine dysregulation
has proven to be quite complex and the answer may not prove to
and TSC/mTOR/HIF/VEGF activation may be a general
be straightforward. Nonetheless, our results collectively suggest
mechanism linking KS co-factors, inflammation, and dysregu-
that there is a redundancy in these pathways and in the
lated angiogenesis in KS.
phosphorylation sites for inhibiting TSC activity and further
The mTOR signaling pathway is a key modulator of protein
provide insight into the complexity of mTOR regulation by
translation and has previously been identified as a positive
different exogenous stimuli.
regulator of HIF and HIF-dependent responses [20,30].
A number of vGPCR factors promote the phosphorylation of
Oncogenes activating this pathway have been implicated in
TSC1/2 through several of these signaling pathways. Surprisingly,
tumor-induced angiogenesis in other tumors. vGPCR promo-
we demonstrate here that phosphorylation of TSC1 in Ser511 by
tion of the paracrine activation of mTOR may play a similar
IKKb is activated by most of the cytokines tested (Fig. S1),
role in the regulation of HIF in KS. Indeed, upregulation of
suggesting that regulation of mTOR by IKKb may be quite
HIF activity has been observed upon KSHV infection of
promiscuous. Moreover, as IKKb activation leads to the activation
endothelial cells in culture and HIF stabilization has been
of NFkB, this, in turn, may promote a positive feedback loop,
previously reported in AIDS-KS lesions [40,41]. Of note, other
further enhancing cytokine – and therefore VEGF – secretion in
KSHV genes (e.g. LANA-1 and IRF-3) have also been
KS. Of note, paracrine secretions from vGPCR-expressing cells
suggested to play a role in upregulating HIF activity
promote a gene expression profile with an NFkB signature in
[42,43,44]. Given the central role of HIF in VEGF regulation,
endothelial cells [32]. In light of our results here, this suggests that
and the importance of VEGF in KS, it is certainly reasonable to
the NFkB signature may be mediated by the specific secreted
argue that KSHV may encode a redundancy of mechanisms to
Figure 7. vGPCR cytokines activate VEGF secretion through diverse signaling cascades converging in TSC/mTOR/HIF. Schematicshowing the different signaling pathways by which vGPCR cytokines, chemokines and growth factors converge in the phosphorylation of TSC1/2, theactivation of mTOR and the upregulation of HIF levels, leading to VEGF secretion.
doi:10.1371/journal.pone.0019103.g007
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
ensure that this critical endothelial cell growth factor is available
thermocycler from eppendorfs (2 min at 94uC; 30 cycles of 94uC
in growing KS tumors.
for 30 seconds, 50uC for 30 seconds, 72uC for 1 minute; and
Here, we show that pharmacological inhibition of HIF
5 minutes at 72uC). Primers for human VEGF were: 59-GG-
upregulation by vGPCR is sufficient to inhibit vGPCR oncogen-
GCAGAATCATCACGAAGT-39 (sense) and 59-TGGTGAT-
esis. As the master regulator of the hypoxic vascular response, it
GTTGGACTCCTCA-39 (anti-sense). Primers for human HIF-
should not be surprising that HIF plays a central role in Kaposi's
1a were: 59-GCAAGCCCTGAAAGCGCAAG-39 (sense) and 59-
sarcomagenesis. HIF drives transcriptional activation of hundreds
GTGAGGCTGTCCGACTTTGA-39 (anti-sense). For HIF-2a,
of genes involved in vascular reactivity, angiogenesis, arteriogen-
primers employed were: 59-GTCTCTCCACCCCATGTCTC-39
esis, and the recruitment of endothelial precursor cells, all key steps
(sense) and 59-GGTTCTTCATCCGTTTCCAC-39 (anti-sense).
toward the development of KS [45]. Indeed, it is tempting to
Amplification of GAPDH sequence was used for normalization.
speculate that recent publications describing KS regression inpatients with iatrogenic KS following a switch in their immuno-
Western blots and Immunohistochemistry
suppressive treatment to the mTOR inhibitor, Sirolimus, may – in
Western blots and immunohistochemistry were performed as
part – be due to its effect on decreasing HIF activation [46,47].
previously described [12]. Antibodies recognizing AU1, AU5 and
Collectively, our data ultimately provide the basis for the early
HA epitopes were purchased from Covance. Antibodies against
assessment of drugs inhibiting HIF in those patients with
the following proteins were employed: S6K, p-S6K (T389), S6,
cutaneous and/or systemic KS.
p-S6 (S240/244), AKT, TSC2, p-TSC2 (T1462), ERK1/2,IKKb, p-IKK-a/b (S180/181), mTOR, p-mTOR (S2448), p38,
Materials and Methods
p-p38 (T180/Y182), HIF-1a and HIF-1b from Cell Signaling;
Expression plasmids and reagents
HIF-2a and P-TSC2 (S664) (immunohistochemistry) from NovusBiologicals; TSC1 from Invitrogen; p-TSC1 (S511) from Bethyl
The expression plasmid encoding for the rapamycin-resistant
Laboratories; LANA-1 from Leica Microsystems; p-AKT (S473)
mTOR mutant (RR-mTOR) that bears a Ser2035RIle (SI)
from R&D Systems; p-ERK1/2 (T202/Y204) from BD Biosci-
substitution in the FKBP12-rapamycin-binding domain has been
ences; and Actin from Santa Cruz. For p-TSC2 (S1254),
described elsewhere [19]. A tetracycline inducible system (Tet-on)
antibodies from Enogene and Cell Signaling were used for
was used for vGPCR expression. pCEFL Tet REV TA and pBIG
Western blot and immunohistochemistry, respectively. For VEGF,
AU5 vGPCR were kindly provided by Dr. Silvio Gutkind(NIDCR, NIH). GRO-a was obtained from R&D Systems; IL-8,
antibodies from R&D Systems and Abcam were used for Western
VEGF, PDGF, IL-1b, IL-10, IL-6, TNFa, IP-10 and SDF-1a
blot and immunohistochemistry, respectively. The antibody
were obtained from Peprotech. Rapamycin, LY294002, U0126,
recognizing vGPCR was kindly provided by Dr. Gary S. Hayward
SB203580 and BAY 117082 were purchased from Calbiochem
(Johns Hopkins University, Baltimore, MD).
and Digoxin from Sigma. All siRNA oligos were obtained fromQiagen.
Tumorigenesis assays
All procedures involving animals were approved by the
Cell lines, transfections and supernatant collection
Institutional Animal Care and Use Committee. Murine vGPCR
Immortalized human dermal microvascular endothelial cells
tumors were obtained as described in ref 12. Briefly, TIE2-tva
(HMEC1) were obtained from the Centers of Disease Control
transgenic mice expressing in vascular endothelium the avian
(Atlanta, GA) and grown in Gibco MCDB 131 medium
leukosis virus (ALV) receptor, TVA, were injected i.p. with ALV-
(Invitrogen), supplemented with 10% FBS, 10 mM/l L-Gluta-
derived retrovirus encoding for KSHV vGPCR. Macroscopic
mine, 10 ng/ml epidermal growth factor, 1 mg/ml hydrocortisone
vGPCR tumors developed in 4 months predominantly in ear, tail,
and 1% antibiotic antimycotic. (SV-40) immortalized murine
paw and GI tract. For tumor allograft formation, cells expressing
endothelial cells (SVEC) and SVEC-derived stable cell lines
vGPCR (EC-vGPCR or EC-vGPCR/RR-mTOR) were mixed
expressing KSHV vGPCR or KSHV vCyclin and vFLIP (EC-
with cells expressing vCyclin and vFLIP (EC-vCYC/vFLIP or EC-
vGPCR, EC-vCYC/vFLIP) were described previously [15]. The
vCYC/vFLIP/RR-mTOR), in a (1:10) ratio (105:106 cells). These
Rapamycin-Resistant mTOR (RR-mTOR) was transfected along
mixed cell populations were then injected into the right flank of 8-
with the pTracer-EF/Bsd plasmid (Invitrogen) into EC-vGPCR or
wk-old athymic (nu/nu) nude female mice as previously described
EC-vCYC/vFLIP. Cells were then stably selected with Blasticidin
[15]. For these in vivo studies, Digoxin stock solution (Sigma-
(Invitrogen). siRNA delivery into cultured cells was performed
Aldrich) was dissolved in DMSO and Rapamycin (LC Laborato-
using Hiperfect (Qiagen). For conditioned media preparation,
ries) was dissolved as previously described [19]. Drug treatment
HMEC1 were transfected with Tet REV TA and pBIG AU5
was initiated when estimated tumor weight reached around 150–
vGPCR, serum starved and treated with doxycycline (Dox; 1
200 mg. For this procedure, tumor-bearing animals were
mg/ml). 24 h later, supernatants from untreated (Control CM) and
randomly grouped (control, n = 5; drug-treated group, n = 5) and
Dox treated (vGPCR CM) cells were collected, centrifuged at
treated with Rapamycin (10 mg/kg) or Digoxin (2 mg/kg) or an
1000 g for 10 min, and concentrated 10 times with centrifugal
equal volume of vehicle. For Rapamycin, treatment schedule was
a single injection per animal given i.p. for 5 consecutive days [19].
For Digoxin, treatment schedule was a single injection per animal
Reverse transcriptase PCR
given i.p. for 10 consecutive days [31]. The animals were
Total RNA was isolated using the GenElute Mammalian Total
monitored twice weekly for tumor formation. The longest length
RNA Miniprep kit (Sigma-Aldrich) according to manufacturer's
(L) and shortest width (W) of the tumor were measured using a
protocol and reverse transcription was performed using Super-
caliper at different time points throughout the experiment. Tumor
Script III First-Strand Synthesis System (Invitrogen). Upon
volume was then converted into tumor weight using the formula
extraction of mRNA, cDNA was obtained using the SuperScript
LW2/2, as described previously [19]. Results of animal experi-
III First-Strand Synthesis System from Invitrogen. Subsequently,
ments were expressed as mean estimated tumor weight 6 SD.
the PCR reaction was carried out using the Mastercycler
When appropriate, animals were euthanized, and tissue was fixed
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
in 4% paraformaldehyde and embedded in paraffin for further
S6K and S6 upon treatment of HMEC1 with different vGPCR
(recombinant) proteins: IL-8, GROa, VEGF, PDGF, IL-1b, IL-10, IL-6, TNFa, IP-10, or SDF1a. Cells were pretreated with
(50 nM) Rapamycin, where corresponding. (B) Phosphorylation of
Four patients diagnosed with KS (multiple lesions) at University
TSC2/1 (TSC2T1462, TSC2S664, TSC2S1254 and TSC1S511) or
of Maryland Dental School or School of Medicine, between 1990
S6K upon treatment of HMEC1 with different recombinant
and 2010, were identified. Formalin-fixed paraffin-embedded
cytokines, chemokines, and growth factors, as described in (A). (C)
tissue samples were obtained from the pathology archives for
Analysis of the levels of phosphorylation of TSC2/1 (TSC2T1462,
immunohistochemical studies. Representative hematoxylin and
TSC2S664, TSC2S1254 and TSC1S511) shown in (B).
eosin sections of each tumor were reviewed and the diagnosis was
confirmed by immunohistochemistry using a specific antibodyrecognizing
(LANA-1). The Institutional Review Board (IRB) protocol wasexempt.
We thank Dr. Gary S. Hayward for kindly providing the specific antibodyrecognizing KSHV vGPCR. BCJ is a recipient of a predoctoral fellowshipfrom the CNPq-Brazil. We thank Xin Wang for her help with the
Statistical Analysis
In all cases, results are shown as a mean value 6 SD from at
least three independent experiments. Western blot scans are
Author Contributions
representative of at least three independent experiments. Statisticalanalysis was performed with Prism 4.2 software (GraphPad).
Conceived and designed the experiments: A. Schneider A. Sodhi SM.
ANOVA test: ***, p,0.001; **, p,0.01; *, p,0.05.
Performed the experiments: BCJ TM JH RC ERF. Analyzed the data: BCJTM JH RC ERF SM. Contributed reagents/materials/analysis tools: PPP.
Wrote the paper: SM.
Supporting Information
Specific vGPCR factors induce TSC2/1
phosphorylation at different sites. (A) Phosphorylation of
1. Mesri EA, Cesarman E, Boshoff C (2010) Kaposi's sarcoma and its associated
feration in a murine model of Kaposi's sarcoma. J Immunol 174: 3686–
herpesvirus. Nat Rev Cancer 10: 707–719.
2. Cesarman E, Mesri EA, Gershengorn MC (2000) Viral G protein-coupled
15. Montaner S, Sodhi A, Ramsdell AK, Martin D, Hu J, et al. (2006) The Kaposi's
receptor and Kaposi's sarcoma: a model of paracrine neoplasia? J Exp Med 191:
sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic
target for the treatment of Kaposi's sarcoma. Cancer Res 66: 168–174.
3. Cornali E, Zietz C, Benelli R, Weninger W, Masiello L, et al. (1996) Vascular
16. Jham BC, Montaner S (2010) The Kaposi's sarcoma-associated herpesvirus G
endothelial growth factor regulates angiogenesis and vascular permeability in
protein-coupled receptor: Lessons on dysregulated angiogenesis from a viral
Kaposi's sarcoma. Am J Pathol 149: 1851–1869.
oncogene. J Cell Biochem 110: 1–9.
4. Masood R, Cai J, Zheng T, Smith DL, Naidu Y, et al. (1997) Vascular
17. Sodhi A, Montaner S, Patel V, Zohar M, Bais C, et al. (2000) The Kaposi's
endothelial growth factor/vascular permeability factor is an autocrine growth
sarcoma-associated herpes virus G protein-coupled receptor up-regulates
factor for AIDS-Kaposi sarcoma. Proc Natl Acad Sci U S A 94: 979–984.
vascular endothelial growth factor expression and secretion through mitogen-
5. Nakamura S, Murakami-Mori K, Rao N, Weich HA, Rajeev B (1997) Vascular
activated protein kinase and p38 pathways acting on hypoxia-inducible factor
endothelial growth factor is a potent angiogenic factor in AIDS-associated
1alpha. Cancer Res 60: 4873–4880.
Kaposi's sarcoma-derived spindle cells. J Immunol 158: 4992–5001.
18. Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, et al. (1998) G-
6. Weindel K, Marme D, Weich HA (1992) AIDS-associated Kaposi's sarcoma
protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral
cells in culture express vascular endothelial growth factor. Biochem Biophys Res
oncogene and angiogenesis activator. Nature 391: 86–89.
Commun 183: 1167–1174.
19. Sodhi A, Chaisuparat R, Hu J, Ramsdell AK, Manning BD, et al. (2006) The
7. Brown LF, Tognazzi K, Dvorak HF, Harrist TJ (1996) Strong expression of
TSC2/mTOR pathway drives endothelial cell transformation induced by the
kinase insert domain-containing receptor, a vascular permeability factor/
Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer
vascular endothelial growth factor receptor in AIDS-associated Kaposi's
Cell 10: 133–143.
sarcoma and cutaneous angiosarcoma. Am J Pathol 148: 1065–1074.
20. Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer
8. Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, et al. (2003) Kaposi's
Cell 12: 9–22.
sarcoma associated herpesvirus G protein-coupled receptor immortalizes human
21. Lee DF, Hung MC (2007) All roads lead to mTOR: integrating inflammation
endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 3:
and tumor angiogenesis. Cell Cycle 6: 3011–3014.
22. Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and
9. Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E (1997)
inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657.
Human herpesvirus KSHV encodes a constitutively active G-protein- coupled
23. Potter CJ, Pedraza LG, Xu T (2002) Akt regulates growth by directly
receptor linked to cell proliferation. Nature 385: 347–350.
phosphorylating Tsc2. Nat Cell Biol 4: 658–665.
10. Cesarman E, Nador RG, Bai F, Bohenzky RA, Russo JJ, et al. (1996) Kaposi's
24. Dan HC, Sun M, Yang L, Feldman RI, Sui XM, et al. (2002) Phosphatidy-
sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin
linositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor
D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma.
complex by phosphorylation of tuberin. J Biol Chem 277: 35364–35370.
J Virol 70: 8218–8223.
25. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005)
11. Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, et al. (2003)
Phosphorylation and functional inactivation of TSC2 by Erk implications for
Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic
tuberous sclerosis and cancer pathogenesis. Cell 121: 179–193.
mouse. J Virol 77: 2631–2639.
26. Li Y, Inoki K, Vacratsis P, Guan KL (2003) The p38 and MK2 kinase cascade
12. Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, et al. (2003)
phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its
Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates
interaction with 14-3-3. J Biol Chem 278: 13663–13671.
Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral
27. Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, et al. (2007) IKK beta
latent genes. Cancer Cell 3: 23–36.
suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR
13. Yang TY, Chen SC, Leach MW, Manfra D, Homey B, et al. (2000) Transgenic
pathway. Cell 130: 440–455.
expression of the chemokine receptor encoded by human herpesvirus 8 induces
28. Montaner S, Sodhi A, Servitja JM, Ramsdell AK, Barac A, et al. (2004) The
an angioproliferative disease resembling Kaposi's sarcoma. J Exp Med 191:
small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to
cytokine secretion and paracrine neoplasia. Blood 104: 2903–2911.
14. Jensen KK, Manfra DJ, Grisotto MG, Martin AP, Vassileva G, et al. (2005) The
29. Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism.
human herpes virus 8-encoded chemokine receptor is required for angioproli-
Curr Opin Genet Dev 20: 51–56.
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
VEGF Amplification by vGPCR Activation of HIF
30. Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer
39. Rosenberg ZF, Fauci AS (1988) Immunopathogenic mechanisms in human
biology and therapeutics. Oncogene 29: 625–634.
immunodeficiency virus (HIV) infections. Ann N Y Acad Sci 546: 164–174.
31. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, et al. (2008) Digoxin and other
40. Carroll PA, Kenerson HL, Yeung RS, Lagunoff M (2006) Latent Kaposi's
cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc
sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-
Natl Acad Sci U S A 105: 19579–19586.
induced factors. J Virol 80: 10802–10812.
32. Martin D, Galisteo R, Ji Y, Montaner S, Gutkind JS (2008) An NF-kappaB gene
41. Catrina SB, Botusan IR, Rantanen A, Catrina AI, Pyakurel P, et al. (2006)
expression signature contributes to Kaposi's sarcoma virus vGPCR-induced
Hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha are
direct and paracrine neoplasia. Oncogene 27: 1844–1852.
expressed in kaposi sarcoma and modulated by insulin-like growth factor-I.
33. Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, et al. (2002) The human
Clin Cancer Res 12: 4506–4514.
herpes virus 8-encoded viral FLICE inhibitory protein physically associates with
42. Cai Q, Murakami M, Si H, Robertson ES (2007) A potential alpha-helix motif in
and persistently activates the Ikappa B kinase complex. J Biol Chem 277:
the amino terminus of LANA encoded by Kaposi's sarcoma-associated
herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia.
J Virol 81: 10413–10423.
34. Sun Q, Matta H, Lu G, Chaudhary PM (2006) Induction of IL-8 expression by
43. Cai QL, Knight JS, Verma SC, Zald P, Robertson ES (2006) EC5S ubiquitin
human herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene
complex is recruited by KSHV latent antigen LANA for degradation of the
25: 2717–2726.
VHL and p53 tumor suppressors. PLoS Pathog 2: e116.
35. Sakakibara S, Pise-Masison CA, Brady JN, Tosato G (2009) Gene regulation
44. Shin YC, Joo CH, Gack MU, Lee HR, Jung JU (2008) Kaposi's sarcoma-
and functional alterations induced by Kaposi's sarcoma-associated herpesvirus-
associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible
encoded ORFK13/vFLIP in endothelial cells. J Virol 83: 2140–2153.
factor-1 alpha to induce vascular endothelial growth factor expression. Cancer
36. McCormick C, Ganem D (2005) The kaposin B protein of KSHV activates the
Res 68: 1751–1759.
p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307: 739–741.
45. Rey S, Semenza GL (2010) Hypoxia-inducible factor-1-dependent mechanisms
37. Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, et al. (2003)
of vascularization and vascular remodelling. Cardiovasc Res 86: 236–242.
Activation of mitogen-activated protein kinase and NF-kappaB pathways by a
46. Campistol JM, Gutierrez-Dalmau A, Torregrosa JV (2004) Conversion to
Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol 77:
sirolimus: a successful treatment for posttransplantation Kaposi's sarcoma.
Transplantation 77: 760–762.
38. Tomlinson CC, Damania B (2004) The K1 protein of Kaposi's sarcoma-
47. Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, et al. (2005) Sirolimus
associated herpesvirus activates the Akt signaling pathway. J Virol 78:
for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med 352:
PLoS ONE www.plosone.org
April 2011 Volume 6 Issue 4 e19103
Source: http://www.enogene.com.cn/uploadfile/Amplification%20of%20the%20Angiogenic%20Signal%20through%20the.pdf
IL VALORE SOCIALE ED Assessorato al volontariato ed alla sanità NOMICO DEL VOLONTARIATO. UNA RICERCA C Il volume presenta i principali risultati di una ricerca condotta nella regione Marche sul tema del valore IL VALORE SOCIALE ED ECONOMICO sociale ed economico del volontariato, promossa dall'Associazione volontariato Marche - Centro servizi per il volontariato, in collaborazione con l'Università di Urbino "Carlo Bo" e la Regione Marche.
Gurnee Police Department Honor • Integrity • Service CONTENTS The Cover Photo On June 15th, a 911 Award was presented to 9 year old Angel Escobar for calling 911 for his mother in need of medical attention. The incident occurred on May 10th. Angel was able to give their address and talk with the telecommunicator in a very stressful situation. Angel,


