
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Nuclear permeable ruthenium(ii) -carboline complexes induce autophagy to antagonize mitochondrial-mediated apoptosis
J. Med. Chem. 2010, 53, 7613–7624 7613
DOI: 10.1021/jm1009296
Nuclear Permeable Ruthenium(II) β-Carboline Complexes Induce AutophagyTo Antagonize Mitochondrial-Mediated Apoptosis
Caiping Tan,† Sensen Lai,† Shouhai Wu,† Sheng Hu,§ Lingjun Zhou,† Yu Chen,† Minxu Wang,† Yiping Zhu,†Wu Lian,† Wenlie Peng,† Liangnian Ji,‡ and Anlong Xu*,†
†State Key Laboratory of Biocontrol, Department of Biochemistry, College of Life Sciences, Sun Yat-sen University, Guangzhou,Guangdong 510006, P. R. China, ‡MOE Laboratory of Bioinorganic and Synthetic Chemistry,School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China, and
§Faculty of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China
Received July 22, 2010
The role of autophagy in cancer development and response to cancer therapy has been a subject of debate.
Here we demonstrate that a series of ruthenium(II) complexes containing a β-carboline alkaloid as ligandcan simultaneously induce autophagy and apoptosis in tumor cells. These Ru(II) complexes are nuclearpermeable and highly active against a panel of human cancer cell lines, with complex 3 displaying activitiesgreater than those of cisplatin. The antiproliferative potentialities of 1-3 are in accordance with theirrelative lipophilicities, cell membrane penetration abilities, and in vitro DNA binding affinities. Com-plexes 1-3 trigger release of reactive oxygen species (ROS) and attenuation of ROS by scavengers reducedthe sub-G1 population, suggesting ROS-dependent apoptosis. Inhibition of ROS generation also reducesautophagy, indicating that ROS triggers autophagy. Further studies show that suppression of autophagyusing pharmacological inhibitors (3-methyladenine and chloroquine) enhances apoptotic cell death.
mitochondrial pathway in otherwise highly chemoresistantmelanoma cells.11
Autophagy, or type II programmed cell death, has been
Previously, a number of ruthenium compounds have been
proposed as a third mode of cell death besides apoptosis and
shown to display promising anticancer activity. Two Ru(III)
necrosis.1,2 Autophagy is a double-edged sword in oncology,
complexes have successfully entered clinical trials, namely,
as it is involved in both cell survival and cell death. It is long
known to provide a survival advantage to rapidly growing cells
4(DMSO)(Im)], where Im =
imidazole and DMSO = dimethylsulfoxide)a and KP101913
under conditions of hypoxic or metabolic stresses, which thus
contributes to normal and cancer cell survival. It also has a role
4(Ind)2], where Ind = indazole). Ru(II)
complexes carrying labile ligands such as "half-sandwich"
in the suppression of tumor growth, and autophagy defects are
arene complexes exhibit both in vitro and in vivo activities.14,15
associated with increased tumorigenesis.3,4 The role of autop-
Some of the R-[Ru(II)(azpy)
hagy in cancer therapy is also a topic of intense debate, and
2Cl2] (azpy = 2-(phenylazo)-
pyridine) type complexes show a cytotoxic potency similar
autophagy can serve as a cell survival pathway by suppressing
to or even better than that of cisplatin.16 It has also been
apoptosis; thus, treating cancer cells by autophagy inhibition is
reported that coordinatively saturated Ru(II) polypyridyl
possible.5 It can also lead to death itself; thus, drugs can
activate autophagy to kill cancer cells resistant to apoptosis.6
2(dppn)]Cl2 (dppn = 4,5,9-16-tetraazadi-
benzo[a,c]naphthacene) exhibits cytotoxic activity against
As autophagy is such a fundamental process, establishing how
two cancer cell lines at low micromolar IC
the functional status of autophagy influences the response to
The β-carboline alkaloids are a class of synthetic and
cancer treatment is important.4,7
naturally occurring compounds that possess a large spectrum
The development of metal complexes with bioactive mole-
cules as ligands offers possibilities for the discovery of novelanticancer drugs with enhanced and targeted activity.8 Sub-
a Abbreviations: 1-Py-βC, 1-(2-pyridyl)-β-carboline; 3-MA, 3-methy-
stitution of the β-phenyl ring of tamoxifens by ferrocenyl
ladenine; AO, acridine orange; AVOs, acidic vesicular organelles; azpy,
group affords ferrocifens, which exhibit a strong antiprolifera-
2-(phenylazo)pyridine; bpy, 2,20-bipyridine; CCCP, carbonyl cyanidem-chlorophenylhydrazone; CDKs, cyclin-dependent kinases; CQ,
tive effect in hormone-independent breast cancer cells, where
chloroquine; DIP, 4,7-diphenyl-1,10-phenanthroline; CT-DNA, calf
tamoxifen and ferrocene are inactive.9 The combination of
thymus DNA; DMSO, dimethylsulfoxide; dppn, 4,5,9-16-tetraazadi-
Ru(II) arene complexes with staurosporine, a potent inhibi-
benzo[a,c]naphthacene; dppz, dipyrido[3,2-a:20,30-c]phenazine; GF-AAS, graphite furnace atomic absorption spectrometry; H
tor for various kinases, results in the discovery of nanomolar
20,70-dichlorofluorescein diacetate; Im, imidazole; Ind, indazole; JC-1,
and even picomolar protein kinase inhibitors,10 and nota-
bly, the GSK-3β inhibitor DW1/2 is highly cytotoxic
dide; MLCT, metal-to-ligand charge transfer; MMP, mitochondrial
in vitro and can induce p53-activated apoptosis via the
membrane potential; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide; NAC, N-acetylcysteine; PBS, phosphate bufferedsaline; phen, 1,10-phenanthroline; ROS, reactive oxygen species; TEM,
*To whom correspondence should be addressed. Phone/Fax: 86-20-
transmission electron microscopy; Tiron, 4,5-dihydroxy-1,3-benzenedi-
3933-2990. E-mail:
[email protected].
sulfonic acid disodium salt.
r 2010 American Chemical Society
Published on Web 10/19/2010
7614 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
ROS (reactive oxygen species) mediated mechanism. Autop-hagy caused by 1-3 is detected by TEM (transmissionelectron microscopy) and analysis of the localization of greenfluorescent protein-tagged autophagic marker microtubule-associated protein light chain 3 (GFP-LC3) in transfectedHeLa cells. Inhibition of autophagy by pretreatment with3-methyladenine (3-MA) or chloroquine (CQ) sensitizes thecells to apoptotic cell death, revealing that Ru(II)-inducedautophagy plays a cytoprotective role. Additionally, confocalmicroscopy studies show that 3 can penetrate the nuclearenvelope, making it possible that the primary target of thesecomplexes is genomic DNA, so the binding of 1-3 with DNAhas been studied by absorption and fluorescence spectro-scopic studies to further elucidate whether the mechanism of1-3 involves direct DNA damage.
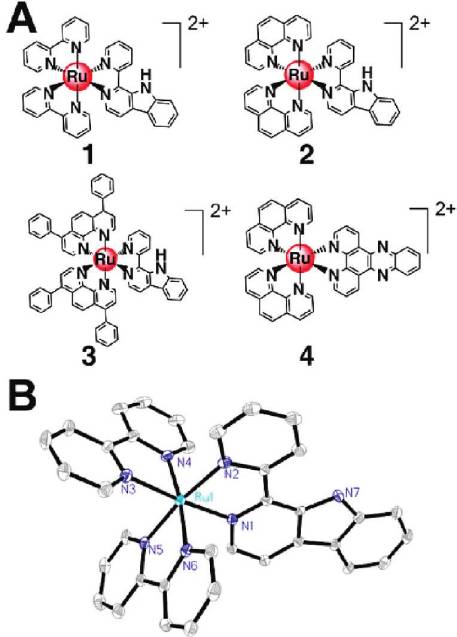
Synthesis and Characterization. The synthesis of the com-
plexes [Ru(N-N)2(1-Py-βC)](PF6)2 (1-3) was carried out asfollows: (i) Synthesis of the ligand 1-Py-βC was achieved byreacting tryptamine and 2-pyridinecarboxaldehyde in dryanisole; (ii) [Ru(N-N)2(1-Py-βC)](PF6)2 (1-3) were syn-thesized by refluxing 1 equiv of 1-Py-βC and cis-[Ru-(N-N)2Cl2] 3 2H2O in 75% (v/v) aqueous ethanol for >3 h,
followed by anion exchange with NH4PF6, purification bycolumn chromatography, and recrystallization. The com-plexes were obtained as racemic mixtures containing bothΔ- and Λ-isomers. The ligand and the complexes were charac-
Figure 1. (A) Structures of the Ru(II) complexes. (B) ORTEP view
terized by 1H NMR spectroscopy, ESI-MS (Supporting
of 1 (30% probability ellipsoids). Hydrogen atoms, solvated molec-
Information Figures S2-S9), and elemental analysis.
ules, and anions are omitted for clarity.
Complexes 1-3 show intense spin-allowed intraligand
(1IL) absorption bands in the UV region at approximately
of important pharmacological properties including sedative,
250-340 nm and less intense spin-allowed metal-to-ligand
anxiolytic, hypnotic, anticonvulsant, antitumor, antiviral,
charge transfer (1MLCT) absorption bands at approxi-
antiparasitic, and antimicrobial activities.18 It has been re-
mately 350-530 nm (Supporting Information Figure S10),
ported that β-carboline alkaloids can exert antitumor activ-
which are typical absorption properties of Ru(II) polypyr-
ities through multiple mechanisms, such as intercalating into
idine complexes. 1-3 display luminescence (550-700 nm) in
DNA19 and inhibiting topoisomerases I and II,20 CDKs (cyclin-
Tris-HCl buffer at 298 K upon excitation at 488 nm. The
dependent kinases),21 and IκB kinases.22 Various synthetic
emission maxima of all the complexes occur at approxi-
derivatives with different substituents in positions 1, 3, and 9
mately 604-609 nm (Figure 9B). The emission is attributed
of the β-carboline skeleton have been synthesized in order to
to a 3MLCT (dπ(Ru) f π*(diimine)) excited state.25 The
elucidate the structure-activity relationship of this class of
DIP complex 3 displays more intense emission than 1 and 2
compounds.18 Al-Allaf and co-workers have reported the
do, and the reason is that the π* orbitals of DIP are lying in
synthesis and the preliminary biological results of some Pd(II)
lower energy than bpy and phen, owing to the electron-
and Pt(II) complexes based on monodentate β-carboline
withdrawing phenyl substitutions.26
alkaloids.23 The Pd(II) complex trans-[Pd(DMSO)(harmine)-
The X-ray crystal structures of 1-Py-βC and complex 1
Cl2] shows a potency better than those of cisplatin, fluorour-
are depicted in Figure S1 (Supporting Information) and
acil and carboplatin, while the mechanism of antitumor action
Figure 1B, respectively. The crystallographic data and se-
by these complexes still remains largely speculative.24
lected bond lengths and angles are listed in Tables S1 and S2
The objective of the present study is to investigate the
(Supporting Information), respectively. The Ru(II) center of
potential of ruthenium β-carboline complexes as anticancer
complex 1 adopts a distorted octahedral geometry, and the
drugs. Three luminescent Ru(II) complexes, [Ru(N-N)2-
average Ru-N bond length is 2.059 A˚, which is comparable
(1-Py-βC)](PF6)2 (N-N = 2,20-bipyridine (bpy, 1), 1,10-
to those found for [Ru(bpy)3]2þ (2.056 A˚) and related com-
phenanthroline (phen, 2), 4,7-diphenyl-1,10-phenanthroline
pounds.27 The average bite angle of bpy (87.10(13)°) falls
(DIP, 3); 1-Py-βC = 1-(2-pyridyl)-β-carboline) (Figure 1A),
within the same range as those of related tris(diimine)-
have been synthesized and characterized. [Ru(phen)2-
(dppz)](PF6)2 (4, dppz = dipyrido[3,2-a:20,30-c]phenazine),
Cellular Uptake. The cellular uptake properties of metal-
which lacks a β-carboline moiety compared with 2, is used
based anticancer drugs are important factors that can influ-
as a reference in biological assays to investigate whether the
ence their antiproliferative efficacies. For platinum com-
biological activities of these complexes are associated with the
plexes, increasing lipophilicities enhances the rate of cellular
β-carboline moiety. We show for the first time that Ru(II)
uptake and, consequently, the cytotoxic activities.29 The
complexes can induce autophagy and apoptosis simulta-
cellular uptake characteristics of Ru(II) complexes can
neously in HeLa (human cervical cancer) cells through a
be estimated by their lipophilicities, which are commonly
Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21 7615
referred to as the n-octan-1-ol/water partition coefficients
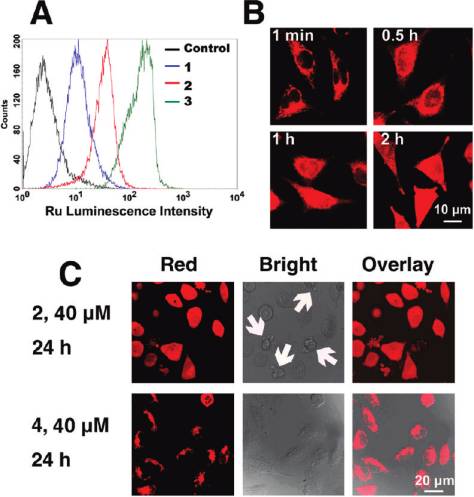
flow cytometry was used to obtain semiquantitative data on
(expressed as log Po/w).26,30 The log Po/w values of 1-3 were
the uptake of Ru(II) complexes into HeLa cells (Figure 2A).
determined by reversed-phase HPLC (Supporting Infor-
Cells not treated with Ru(II) exhibit neglectable background
mation). The lipophilicities of the complexes are substan-
luminescence. Incubation with 20 μM complexes for 2 h
tially increased by incorporating more hydrophobic ligand
causes changes in the luminescence profiles in the following
DIP, and the log Po/w value of the DIP complex 3 (3.39) is
order: 3 > 2 > 1. The luminescence intensity of the cell
much larger than those of 1 (0.52) and 2 (1.43). The high
population increases dramatically after cells are incubated
lipophilicity of the DIP complex is anticipated to facilitate
for 2 h with 20 μM 3. The time-dependent uptake profiles of
the tissue and cellular uptake.
1-3 have also been measured (Supporting Information
Because of their stability in aqueous solution and lumines-
Figure S11). The complexes have accumulated in HeLa cells
cence (the luminescence intensity is increased upon binding
in 30 min. In addition, either lowering the incubation tem-
with DNA, Figure 9B), the cellular uptake properties of
perature or depleting the cellular ATP with CCCP (carbonyl
Ru(II) polypyridyl complexes can be studied using flow cyto-
cyanide m-chlorophenylhydrazone) shows little influence
metry and confocal microscopy conveniently.26,30,31 First,
on uptake (Supporting Information Figure S12), which leadsus to conclude that uptake might occur via an energy-independent process, i.e., passive diffusion, as proposed for[Ru(DIP)2(dppz)]Cl2.31
Flow cytometry cannot discriminate among membrane-
associated, cytoplasmic, and nuclear localization, while thecellular localization characteristics of anticancer drugs arefundamental to their efficacy,32,33 thus the cellular distribu-tion Ru(II) has been studied by confocal microscopy. Inter-estingly, unlike [Ru(DIP)2(dppz)]Cl2, of which almost nonuclear staining can be observed,26 complex 3 (5 μM) graduallypenetrates into the interior of the nucleus and shows diffusecytoplasmic and nuclear fluorescence after 2 h of incubationwith HeLa cells (Figure 2B). The impact of the incorporationof 1-Py-βC on cellular distribution of Ru(II) complexes hasbeen further investigated by using 4 as a reference, whichlacks the β-carboline moiety. After incubation at 40 μM for24 h, 2 produces a diffuse cytoplasmic and nuclear fluorescencein HeLa cells. In contrast, 4 accumulates in the cytoplasmand is predominantly excluded from the nucleus. Notably,cells treated with 2 show marked morphological changes(e.g., cell shrinkage and membrane blebbing).
The cellular ruthenium content of the samples was also
determined by GF-AAS (graphite furnace atomic absorptionspectrometry) quantitatively using the literature method.17
Figure 2. (A) Flow cytometric results of HeLa cells incubated with
The protein content was determined by the Bradford meth-
blank medium and complexes 1 (20 μM), 2 (20 μM), and 3 (20 μM) at37 °C for 2 h. 10 000 events were collected in the FL2 channel
od, and results were reported as ng of ruthenium per mg of
(excitation, 488 nm; emission, 585 ( 21 nm). (B) Confocal images of
cellular protein. The uptake levels are relatively low for
HeLa cells treated with 3. Cells were incubated with 5 μM complex 3
complexes 1 (148.1 ( 20.5) and 2 (173.9 ( 15.6). An incuba-
for different time intervals at 37 °C and observed by confocal
tion concentration of 500 μM has to be used for 1 and 2 in
microscopy (excitation, 488 nm; emission, 600-630 nm). (C) Com-
order to reach detectable cellular Ru level, while 3 (103.1 (
parison of the cellular localization of 2 and 4 taken by confocal
5.8) shows a comparable Ru level after exposure to a much
microscopy (excitation, 488 nm; emission, 600-630 nm). Note themorphological changes (cell shrinkage and membrane blebbing)
lower concentration (10 μM). The cellular uptake data ob-
caused by treatment of 2 (indicated by arrows).
tained for complexes 1-3 show a clear correlation between
Table 1. IC50 Values of Tested Compounds Towards Different Cell Linesa
a IC50 values are given in μM, and those of 1-Py-βC, the Ru(II) precursors, 4, NAMI-A, and cisplatin are included for comparison. Data are presented
as mean values ( standard deviations, and cell viability is assessed after 48 h of incubation.
7616 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
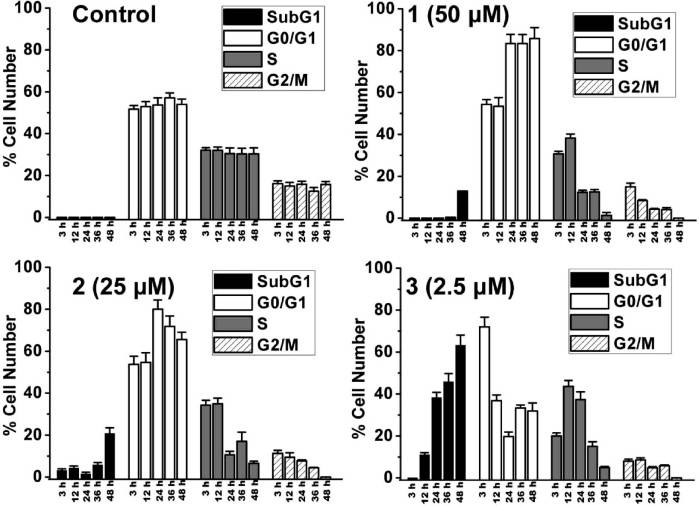
Figure 3. Quantitative cell cycle distribution data for HeLa cells after treatment with 1-3. Data shown are mean values ( standard deviationsof three samples for each treatment.
the lipophilicity and the amount of ruthenium associated
and 2 cause an increase of the sub-G1 fraction (0.8 ( 0.1%,
with the cells.
12.9 ( 0.2%, and 20.7 ( 2.8% for control, 1, and 2,
In Vitro Cytotoxicity. To explore the antitumor potential
respectively), an index of apoptotic DNA fragmentation.
of the Ru(II) complexes, HepG2 (human hepatocellular
Notably, 3 (2.5 μM) causes a significant and time-dependent
liver carcinoma), HeLa, MCF-7 (human breast adeno-
increase in the percentage of cells in sub-G1 phase. After 48 h
carcinoma), and MCF-10 (immortalized human breast
of incubation, most of the cells treated with 3 (63.2 ( 4.9%)
epithelial) cells were treated with varying concentrations
have undergone apoptosis.
of Ru(II) for 48 h, and cell viability was determined by the
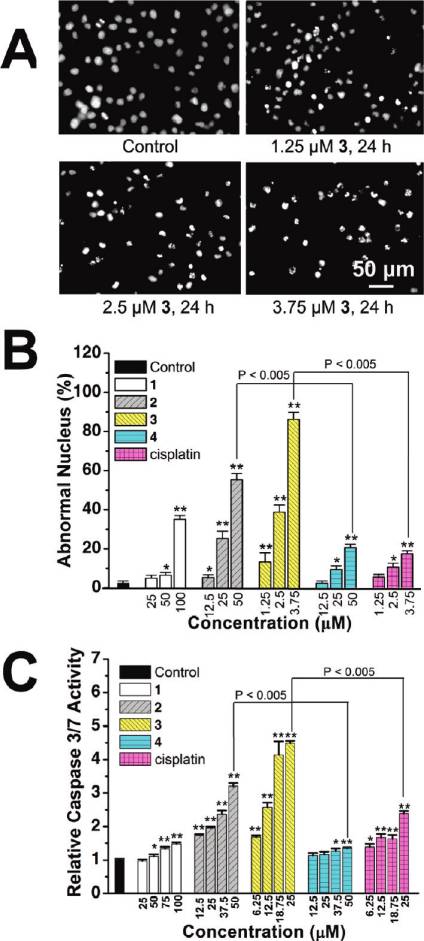
To observe the morphologic characteristics of apoptotic
nuclei, HeLa cells were stained with Hoechst 33342 after
lium bromide) assay. 1-Py-βC, three Ru(II) synthetic pre-
exposure to serial concentrations of 1-4 and cisplatin for
cursors, complex 4, NAMI-A, and cisplatin were included
24 h and detected by fluorescence microscopy. Representa-
as controls. The resulting IC50 values for the tested com-
tive images of the cells treated with vehicle (1% DMSO) and
pounds are shown in Table 1. The result indicates that, in
3 are shown in Figure 4A. Control cells exhibit homogeneous
general, on the basis of calculated IC50 values, the following
nuclear staining, and apoptotic cells increase gradually in a
order of in vitro antiproliferative activity of the compounds
dose-dependent manner and display typical apoptotic changes
can be considered: 3 > cisplatin > 2 > 1 > 4 > NAMI-A.
(e.g., staining bright, condensed chromatin, and fragmented
The Ru(II) precursors are far more cytotoxic than the
nuclei).34 The percentages of cells showing abnormal nuclei
corresponding Ru(II) β-carboline complexes against all
are shown in Figure 4B, and Ru(II) (1, 100 μM; 2, 50 μM; 3,
the cell lines screened. 1-3 also show an increased cytotoxic
3.75 μM; 4, 50 μM) treatment increases the percentage of
potency if compared with the ligand 1-Py-βC alone, which
abnormal nuclei (1, 35.0 ( 2.0%; 2, 55.3 ( 3.1%; 3, 86.3 (
is inactive against all the cell lines tested (IC50 > 200 μM).
3.5%; 4, 14.3 ( 1.5%) compared with the vehicle-treated
Notably, complex 3 is more potent than cisplatin against all
cells (2.3 ( 1.2%).
the cancer cell lines screened (approximately 9-fold more
The activation of cysteine proteases (caspases) is the best
potent than cisplatin in killing HeLa cells), while its cyto-
recognized biochemical hallmark of both early and late
toxicity against the normal-like breast epithelial cell line,
stages of apoptosis.35 Thus, we determined the effect of Ru(II)
MCF-10 (IC50 ≈ 40.6 μM), is only slightly higher than
treatment on caspase-3/7 activity. HeLa cells were treated
that of cisplatin (IC50 ≈ 56.2 μM). NAMI-A shows a very
with complexes 1-4 and cisplatin at serial concentrations for
low cytotoxic potency, with IC50 values ranging between
6 h, after which caspase-3/7 activity was determined using the
533.5 and 647.3 μM, which is consistent with the literature
Caspase-Glo assay. As shown in Figure 4C, treatment of
HeLa cells with 1-4 and cisplatin results in a concentration-
Cell Cycle Arrest and Induction of Apoptosis. The effect of
dependent increase in caspase-3/7 activity.
1-3 on cell cycle was investigated by flow cytometry in PI
Compared with 4, more apoptotic nuclei, consistent with
(propidium iodide) stained cells after Ru(II) treatment for 3,
higher level of caspase-3/7 activation, are observed when
12, 24, 36, and 48 h (Figure 3 and Supporting Information
HeLa cells are incubated with 2, implying that the β-carbo-
Figure S13). 1 (50 μM) and 2 (25 μM) cause pronounced G0/
line moiety is involved in the cytotoxicity induced by 1-3. It
G1 arrest after 24 h of incubation (83.4 ( 4.3% and 80.0 (
is evident from these studies that 3 displays a significantly
3.4% for 1 and 2, respectively) compared with the vehicle-
higher potency in apoptosis induction than cisplatin does
treated cells (53.7 ( 3.4%). After treatment for 48 h, both 1
under similar conditions.

Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21 7617
Figure 5. Representative TEM images showing the ultrastructureof HeLa cells treated with vehicle (1% DMSO) (A) or 2.5 μM 3 (B-H)for 12 h. Nuclei are labeled N. Autophagic structures including auto-lysosomes (/) and autophagic vacuoles (#) are detected in Ru-treatedcells. Parts C, D, F, and G are pictures with higher magnificationshowing detailed autophagic structures.
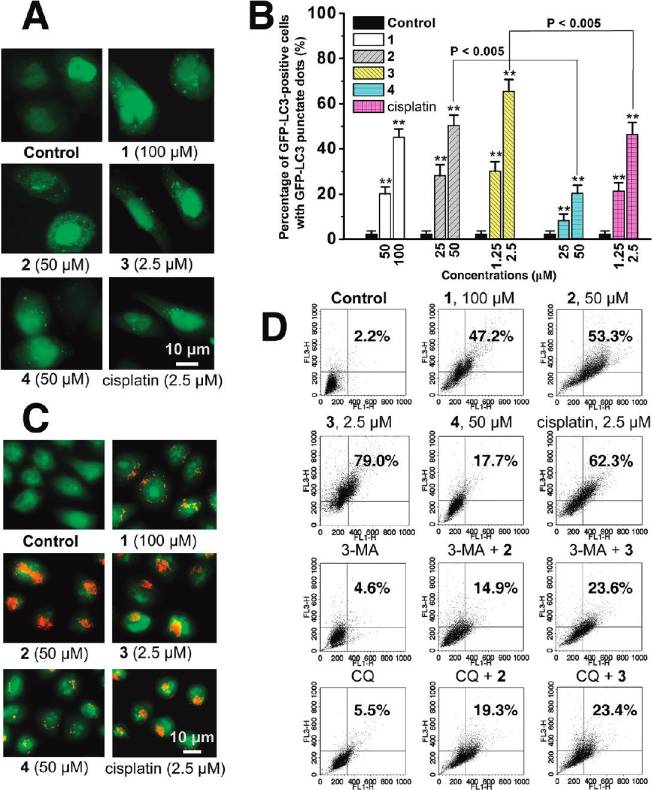
has been examined to determine whether LC3 is concen-trated in HeLa cells after Ru(II) treatment. In cells treatedwith vehicle (1% DMSO), GFP-LC3 protein is diffuselydistributed throughout the cytoplasm, whereas cells treatedwith 1-4 and cisplatin show an increase in number andfrequency of GFP-LC3 dots (g5 dots per cell, Figure 6A).
The percentage of autophagy quantified by counting thenumber of cells showing the punctate pattern of GFP-LC3 in300 GFP-positive cells is shown in Figure 6B, and 65.4 (5.3% of HeLa cells treated with 3 (2.5 μM, 12 h) show GFP-LC3 dots, whereas these autophagic features are detected inonly 2.3 ( 1.3% of cells treated with vehicle.
Autophagy is the process of sequestering cytoplasmic
proteins into lytic components and is characterized by theformation and promotion of AVOs (acidic vesicular organel-les), which can be accessed by AO (acridine orange) stain-
Figure 4. (A) Hochest stained HeLa cells after treatment of 3 at
ing.38,39 As shown in Figure 6C, after Ru(II) treatment for
indicated concentrations after 24 h of incubation. (B) Histogramsshowing the number of cells with abnormal (condensed or fragmented)
12 h, AO staining reveals an increase in the number of cyto-
nuclei. (C) Caspase-3/7 activity after drug treatment for 6 h at
plasmic AVOs, characteristic of autophagy. To quantify the
indicated concentrations. Data shown are mean values ( standard
accumulation of the acidic components, flow cytometry was
deviations from three independent experiments: (/) P < 0.01, (//)
applied to analyze AO-stained cells using the FL3 channel to
P < 0.005, compared with the vehicle-treated cells.
evaluate the bright red fluorescence and the FL1 channel toevaluate the green fluorescence. As shown in Figure 6D,
Autophagy Induced by Ruthenium Complexes. TEM is the
1 (100 μM), 2 (50 μM), and 3 (2.5 μM) treatment increases the
most convincing method for the analysis of autophagy.36
strength of red fluorescence from 2.2% to 47.2%, 53.3%,
Very few autophagical vacuoles are observed, and microvilli
and 79.0%, respectively. Both 3 and cisplatin significantly
are preserved around the cytoplasm in control cells (Figure 5A).
develop AVOs in HeLa cells, and the AVO-inducing ability
Typical autophagic structures, such as autolysosomes and
of 3 is higher than that of cisplatin (62.3%). The formation of
autophagic vacuoles containing cellular material, can be observed
red fluorescent AVOs caused by 2 is more pronounced than
in 3-treated cells (Figure 5B-G). Notably, some of the cells
that caused by 4 (17.7%).
containing autophagic vacuoles simultaneously show mor-
Phosphoinositide 3-kinase (PI3K) is an essential compo-
phologies of apoptosis (apoptotic nuclear condensation and
nent of core machinery involved in autophagic vesicle for-
fragmentation, Figure 5H).
mation. 3-MA, the specific class III PI3K inhibitor, can
One of the hallmarks of autophagy is the conversion of the
inhibit autophagy at an early stage by preventing the forma-
soluble form of LC3 (LC3-I) to the lipidated and autophago-
tion of autophagosomes.40 CQ, a lysosomotropic agent,
some-associated form (LC3-II);37 thus, the presence of a
inhibits autophagy by raising the lysosomal pH, which leads
punctate pattern of GFP-LC3 expression (GFP-LC3 dots)
to inhibition of lysosome-autophagosome fusion and lysosomal
7618 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
Figure 6. (A) Fluorescence staining of GFP-LC3 in response to drug treatment. (B) Autophagosome formation was quantified after 12 htreatment. Data are presented as percentage of GFP-LC3-transfected cells with punctate fluorescence: (/) P < 0.01, (//) P < 0.005, comparedwith vehicle-treated control cells. (C) HeLa cells were stained with AO and examined by fluorescence microscopy after 12 h of treatment.
(D) Quantification of AVOs with AO using flow cytometry. HeLa cells were treated with the complexes for 12 h. For inhibitory experiments, cellswere treated 3-MA (5 mM) or CQ (2.5 μM) for 1 h and further incubated with or without Ru(II) for 12 h. FL1-H indicates green color intensity,while FL3-H shows red color intensity. Top of the grid is considered as AVOs. Data shown are representative of three independent experiments.
protein degradation.41 Preincubation of HeLa cells with these
causes a red to green color shift, indicating loss of MMP, in
inhibitors of autophagy, 3-MA (5 mM) and CQ (2.5 μM),
most of the treated cells. Representative JC-1 red/green ratio
before drug treatment markedly suppresses the induction of
signals recorded by flow cytometry in vehicle-treated (1%
AVOs in HeLa cells (Figure 6D).
DMSO) cells and Ru(II)-treated cells are shown in Figure 7B.
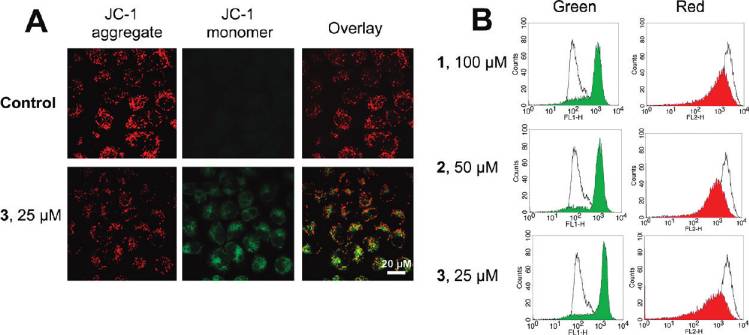
Induction of Mitochondrial Dysfunction. Mitochondrial
After 6 h of incubation, 1 (100 μM), 2 (50 μM), and 3 (25 μM)
dysfunction is involved in both apoptotic and autophagic
treatment decreases the JC-1 red/green ratio signal from 14.5 (
cell death. Mitochondria play important roles in apoptosis
1.1 to 1.2 ( 0.2, 1.0 ( 0.1, and 0.6 ( 0.1, respectively.
through the release of proapoptotic factors such as cyto-
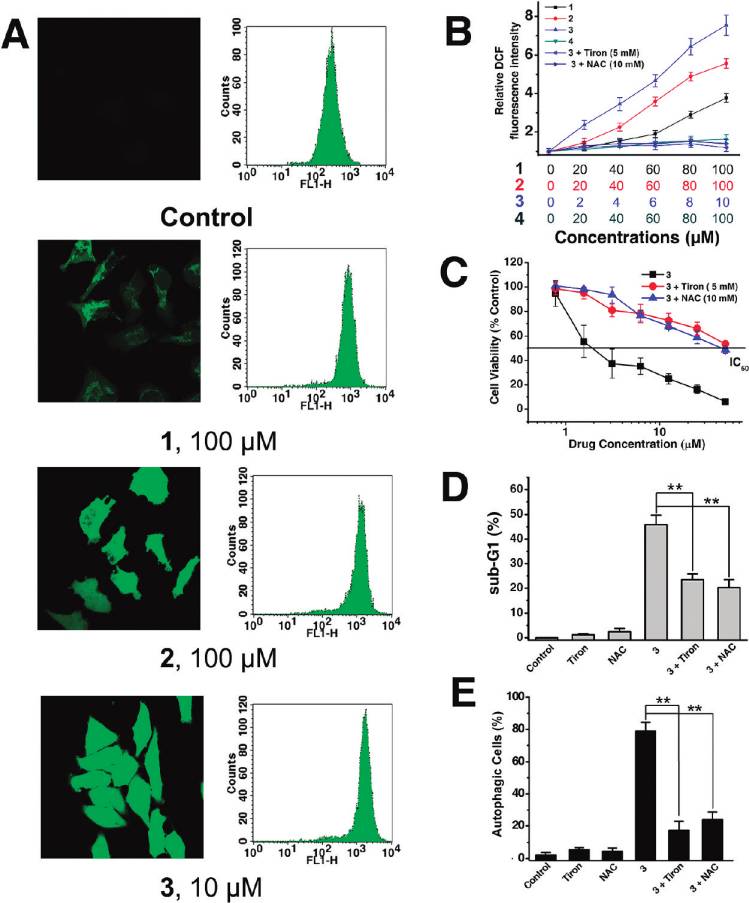
ROS Accumulation. It is well-known that apoptosis can be
chrome c and apoptosis-inducing factor.2,42 As a mechanism
triggered by increased intracellular ROS levels,45 and there is
to maintain genomic integrity in the face of metabolic stress,
strong evidence that ROS are also involved in the induction
drug treatment, or radiation, autophagy can selectively remove
of autophagy.46 Therefore, we investigated whether Ru(II)
damaged mitochondria, which are major sites of genotoxic
treatment could increase the ROS level in HeLa cells. ROS
ROS production.43 Thus, mitochondrial dysfunction was
accumulation was quantified by the 20,70-dichlorofluorescein
determined by measuring changes in the mitochondrial
diacetate (H2DCF-DA) assay. Confocal microscopic anal-
membrane potential (MMP, ΔΨm) using confocal micro-
ysis of DCF-stained Ru(II)-treated cells shows significant
scopy and flow cytometry after staining live cells with the
concentration-dependent increase in intensity of DCF stain-
cationic dye JC-1 (5,50,6,60-tetrachloro-1,10,3,30-tetraethyl-
ing compared with the vehicle-treated cells (Figure 8A, left).
benzimidazolylcarbocyanine iodide). JC-1 exhibits poten-
This result was further confirmed by flow cytometry (Figure 8A,
tial-dependent accumulation in mitochondria, indicated by
right) and microplate analyzer with DCF staining (Figure 8B).
a fluorescence emission shift from red (∼590 nm) to green
3 (10 μM) treatment results in a 7.5-fold increase of fluorescence
(∼525 nm).44 Representative images taken by confocal mi-
signal compared with the vehicle-treated control, at 6 h post-
croscope are shown in Figure 7A. 3 (25 μM, 6 h) treatment
treatment (Figure 8B).
Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21 7619
Figure 7. (A) Fluorescence imaging of JC-1 labeled cells taken by confocal microscope. (B) Effects of 1-3 on MMP analyzed by flowcytometry. Representative histograms of two independent experiments done in triplicate are shown: (unfilled curve) cultures treated withvehicle (1% DMSO); (filled curve) cultures treated with Ru(II).
Having determined that Ru(II) induced generation of
Ru(II) treatment alone. These data therefore further suggest
ROS, we proceeded to examine whether ROS played a role
that Ru(II) complexes stimulate autophagy as a cytoprotec-
in Ru(II)-elicited cell death, autophagy, and apoptosis. We
tive mechanism.
used 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt
DNA Binding Studies. It has been reported that DNA-
(Tiron, 5 mM) and N-acetylcysteine (NAC, 10 mM), two
damaging agents can activate the intrinsic pathway of apop-
ROS scavengers, and examined their effect on cell viability
tosis involving the release of cytochrome c and other mito-
after Ru(II) treatment. Both Tiron and NAC substantially
chondrial apoptogenic factors47 and trigger autophagy
suppress the ROS accumulation (Figure 8B) and reduce the
simultaneously as a self-defense mechanism.48,49 Previously,
cytotoxicity of 3 in HeLa cells (Figure 8C). To investigate the
confocal microscopy studies show that 1-3 can readily pass
involvement of ROS in Ru(II)-induced apoptosis, we mon-
the cell membrane and penetrate into the nucleus (Figure 2),
itored drug-induced sub-G1 population in the absence and
so genomic DNA may serve as the target of these complexes.
presence of Tiron and NAC using flow cytometry. There is a
DNA binding studies have been performed to determine
decline in the drug-induced sub-G1 population when ROS is
whether Ru(II)-induced apoptosis and autophagy are corre-
inhibited by the antioxidants, suggesting that ROS play a
lated with their abilities to cause DNA damage.
crucial role in induction of apoptosis (Figure 8D). Attenua-
The absorption spectra of 1, 2, and 3 in the absence
tion of ROS levels by Tiron and NAC significantly decreases
and presence of CT-DNA (calf thymus DNA) are shown in
the number of AVOs upon Ru(II) treatment (Figure 8E),
Figure 10A. With increasing concentration of CT-DNA,
suggesting ROS also mediate induction of autophagy in
absorption bands of the complexes display clear hypochro-
HeLa cells. Taking all these results together, we conclude
mism. The hypochromism (H% = 100 � (Afree - Abound)/
that ROS have an important role in Ru(II)-induced autop-
Afree) of the 1MLCT bands at ∼470 nm of 1, 2, and 3 are
hagy and apoptosis.
calculated to be about 18.7%, 20.7% and 28.3%, respec-
Inhibition of Apoptosis and Autophagy. To gain better
tively. In order to compare quantitatively the binding affinity
insight into the mechanism of Ru(II) cell-killing action,
of 1-3, the intrinsic binding constants K of 1-3 to DNA
autophagy inhibitors (3-MA and CQ) and caspase inhibitor
were determined by monitoring the changes in absorbance at
(z-VAD-FMK) were used to analyze the interconnection
470 nm (shown in the insets of Figure 10A) using the equation
between Ru(II)-induced cell death, autophagy, and apoptosis.
previously described in the literature.50 The intrinsic binding
First, we determined whether autophagy inhibitors pro-
constants derived for 1, 2, and 3 are (1.08 ( 0.21) � 106 M-1,
moted Ru(II)-induced apoptosis using PI staining and flow
(1.93 ( 0.35) � 106 M-1, and (3.22 ( 0.38) � 106 M-1,
cytometry. Pretreatment with 3-MA (5 mM) promotes Ru-
respectively, which are in the same range as those reported for
(II)-induced apoptosis, which is demonstrated by the in-
Δ-[Ru(phen)2(dppz)]2þ (1.7 � 106 M-1) and Λ-[Ru(phen)2-
creased percentage of sub-G1 phase cells. Similar results
(dppz)]2þ (3.2 � 106 M-1).51 The changes in emission spectra of
are obtained by pretreatment with CQ (2.5 μM) (Figure 9A
1-3 in aqueous solution with increasing DNA concentrations
and Supporting Information Figure S14). These observa-
are shown in Figure 10B. As DNA is successively added, an
tions suggest that Ru(II)-mediated autophagy has a cyto-
obvious increase in emission intensity can be observed. The
protective role in nature.
emission intensities of 1, 2, and 3 increase to about 2.49, 2.97,
Effect of autophagic and caspase inhibitors on Ru(II)-
and 7.48 times of the original intensities, respectively.
induced cell death is shown in Figure 9B. Cells pretreated
Both the electronic absorption titration and fluorescence
with z-VAD-FMK (50 μM) show a marked improvement in
spectroscopic studies indicate that 1-3 have a strong inter-
cell viability relative to cells treated with Ru(II) complexes
action with DNA and the order of DNA binding affinity is
alone, suggesting that Ru(II)-induced cell death is caspase-
3 > 2 > 1. The DNA binding affinities of 1-3 are consistent
dependent, and autophagy may not be an apoptosis-alter-
with their cytotoxicities and potential to induce apoptosis
native pathway to induce cell death. In all cases, the CQ/
and autophagy, implying that DNA may be their primary
Ru(II) and 3-MA/Ru(II) combinations result in reductions
target, and genotoxic stress caused by DNA damage triggers
in cell viability when compared with those observed for
mitochondria-mediated apoptosis and autophagy.
7620 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
Figure 8. Analysis of ROS production after HeLa cells were treated with Ru(II) for 6 h. (A) The intracellular ROS level was detected byconfocal microscope (excitation at 488 nm and emission at 530 nm) and flow cytometry (FL-1 channel; excitation at 488 nm and emission at525 nm). (B) ROS level is expressed as relative fluorescence intensity measured in a microplate reader after 6 h of Ru(II) treatment with orwithout antioxidants at the indicated concentrations. Data are presented as mean values ( standard deviations and are obtained in threeindependent experiments. (C) Effects of Tiron (5 mM) and NAC (10 mM) on 3-induced cytotoxicity. HeLa cells were exposed to different dosesof 3 with or without antioxidants for 48 h. Cell viability was assessed by MTT assay. (D) Attenuating ROS levels by Tiron (5 mM) and NAC(10 mM) reduces the sub-G1 population upon 3 (2.5 μM, 24 h) treatment. (E) Effects of Tiron (5 mM) and NAC (10 mM) on 3-inducedautophagy formation. HeLa cells were exposed to 2.5 μM 3 with or without antioxidants for 12 h. The percentage of autophagic cells wasquantified by AO using flow cytometry as described in Figure 7D: (/) P < 0.01, (//) P < 0.005, compared with the cells treated with 3 alone.
Discussion and Conclusions
space.54 For example, organometallic kinase inhibitors derived
from the class of indolocarbazole alkaloids (e.g., staurosporine)
-Carboline alkaloids are originally isolated from the plant
Peganum harmala. Powdered seeds of Peganum harmala have
can match the size of the active site of targeting kinases and
long been used in herbal formulas of traditional Chinese
discriminate between otherwise closely related binding sites.55
medicine to cure digestive tract tumors.52 Because of their
In the present study, we use the chemically inert and easily
affinity with benzodiazepine, imidazoline, serotonin receptors
synthesized Ru(II) polypyridyl complexes to complement the
of the central nervous system, β-carbolines display considerable
molecular diversity of β-carboline derivatives in the quest for
acute neurotoxicity, which hinders their clinical applications as
the discovery of compounds with superior biological activities
anticancer drugs.18,52 It has long been recognized that, com-
(e.g., improved solubility and selectivity, diminished neurotoxi-
pared with platinum-based anticancer agents, ruthenium com-
city). We present here the first synthesis of a series of ruthenium-
plexes have particularly low general toxicity, allowing admin-
β-carboline complexes that display markedly enhanced cyto-
istration of larger doses.8,53 On the other hand, bulky and rigid
toxicities, compared with the original alkaloid.
octahedral ruthenium complexes can serve as structural scaf-
It has been reported that Ru(II) dipyridophenazine com-
folds to organize the organic ligands in three-dimensional
plexes can readily accumulate in the cytoplasm of live cells but
Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21 7621
are mostly excluded from the nucleus and mainly localized inthe cytoplasm.26,31 For anticancer agents targeting genomicDNA, nuclear accumulation is highly desirable.32,33 Thesubcellular localization of polypyridylruthenium complexescan be greatly influenced by the functional groups (e.g., cell-penetrating peptide,56,57 fluorescein,57 phenanthridine,58 car-boxylic groups,59 and estradiol30) attached to the ligandsystems. It has been reported that β-carboline analogues canpass through the cell membrane after 1 min of drug exposureand enter the nuclear envelope gradually,60 and we speculatethat may partly explain the nuclear penetrating properties ofruthenium β-carboline complexes. A detailed comparison ofcomplexes 2 and 4 indicates that the coordination of the
β-carboline ligand 1-Py-βC to polypyridyl-Ru(II) centersinfluences their cellular localization and results in morecytotoxic complexes. 2 can readily pass through the nuclearenvelope and shows higher ROS, apoptosis, and autophagy-inducing capabilities than 4 does.
Most cancer chemotherapy drugs act through induction of
apoptosis.61 The relationship between autophagy and apop-tosis is complex and depends on particular cell type, stimulus,and environment.62 Evidence has been accumulated to con-firm the importance of autophagy in determining the responseof tumor cells to chemotherapy.63 Under certain circum-stances, autophagy can manifest a cytoprotective role in drugtreatments, such as temozolomide,39 timosaponin A-III,64sulforaphane,65 tamoxifen,66 cisplatin,67 ionizing radiation,68and G-quadruplex ligand.69 Inhibition of autophagy may betherapeutically beneficial in these contexts, as it can sensitize
Figure 9. (A) Apoptosis (formation of sub-G1 peak) was deter-
cancer cells to chemotherapies. Autophagy can also play a
mined after cells were treated with Ru(II) for 48 h at the indicated
destructive role in promoting cell death, as in arsenic
concentrations in the absence or presence of the inhibitors: (/) P <
trioxide-,70,71 resveratrol-,72,73 cannabinoid-,74 and ceramide-
0.01, (//) P < 0.005, compared with the percentage of sub-G1 cells
induced75 autophagy. In these cases, the induction of autop-
for Ru(II) treatment alone. (B) Cell viability was determined by
hagy may be used as a potential therapy for some apoptosis-
MTT assay after cells were treated with 1-3 for 48 h at the indicated
resistant cancers (i.e., breast and pancreatic cancers). For the
concentrations in the absence or presence of 3-MA, CQ, or z-VAD-FMK: (/) P < 0.01, (//) P < 0.005, compared with the cell viability
first time, we find that ruthenium complexes can induce both
of Ru(II) treatment alone.
autophagy and apoptosis in cancer cells. Such dual properties
Figure 10. Absorption spectra of 1-3 in the absence or presence of increasing amounts of CT-DNA. [Ru] = 10 μM, [DNA] = 0-200 μM fromtop to bottom. Arrows indicate the change in absorbance upon increasing the DNA concentration. Inset: plots of (εa - εf)/(εb - εf) vs [DNA]and the nonlinear fit for the titration of the complexes with DNA. Also shown are changes in the emission spectra (λem = 488 nm) of 1-3(10 μM) with increasing concentrations of CT-DNA (0-200 μM).
7622 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
have never been reported for ruthenium complexes or
(Corning). After overnight incubation, the cells were treated
β-carboline derivatives as far as we know. Inhibition of Ru-
with 1 (500 μM), 2 (500 μM), and 3 (10 μM) for 2 h (final DMSO
(II)-induced autophagy by 3-MA or CQ enhances the apop-
concentration, 1% v/v). The cells were trypsinized and washed
tosis-inducing effect with concomitant increased cell death,
twice with cold PBS, and ruthenium content determined by GF-
suggesting that Ru(II)-induced autophagy is a protective
AAS was presented as ng of ruthenium per mg of cellular proteinas described in the literature.17 All experiments were performed
response to the treatment.
in triplicate.
In conclusion, our study shows that the combination of
Autophagy Detection. Transmission Electron Microscopy.
ruthenium complexes with β-carboline alkaloids has a great
HeLa cells (5 � 105) were treated with Ru(II) at 37 °C for 12 h.
potential for developing new anticancer agents. The most
Cells were washed twice and fixed with 2% glutaraldehyde at
active drug, complex 3, exhibits higher cytotoxic potency than
4 °C for 1 h and postfixed with 2% osmium tetroxide. Cells were
the widely used clinical chemotherapeutic agent cisplatin. The
then dehydrated with sequential washes in ethanol and then
antiproliferative effects on tumor cells exerted by 1-3 are
embedded in Spurr's resin. The ultrathin sections obtained were
consistent with their intracellular uptake properties, which are
mounted in copper grids, counterstained with uranyl acetate
significantly enhanced by the increase of lipophilicity with
and lead citrate, and visualized in an electron microscope (JEM
extended π-systems. Further study shows that Ru(II) β-carbo-
100 CX, JEOL, Tokyo, Japan). Images were photographed andscanned by using the Eversmart Jazz program (Scitex).
line complexes exhibit effective cell growth inhibition by
Plasmid and Transfection. Transfection of HeLa cells with
triggering G0/G1 phase arrest and inducing apoptosis through
GFP-tagged LC3 was performed using jetPEI transfection
a ROS-mediated mitochondrial dysfunction pathway. In vitro
reagent (Q-Biogen, France) according to the manufacturer's
DNA binding studies show that genomic DNA may serve as
instructions. Cells were transfected with 1 μg of GFP-LC3
the primary target of these nuclear permeable complexes.
expressing plasmid in each well of 24-well plates. After 12 h,
Notably, these newly synthesized Ru(II) β-carboline com-
cells were treated with 1-3 for 12 h. The cells were then fixed
plexes are dual autophagy- and apoptosis-inducing agents,
with 0.4% paraformalclehyde for 30 min and washed twice with
and they activate autophagy as a cytoprotective response in
PBS. The fluorescence of GFP-LC3 was viewed, and the rate of
HeLa cells, and both the apoptosis- and autophagy-inducing
GFP-LC3 vacuoles was counted under a fluorescent microscope
activities are at least partially related to ROS accumulation.
(Axio Observer Z1, Carl Zeiss, Germany). A minimum of 300GFP-LC3-transfected cells were counted. Cells treated withvehicle solution (1% DMSO) were included as controls.
Experimental Section
Quantification of AVOs with AO. HeLa cells were cultured
Materials and General Methods. All solvents were of analy-
with medium containing vehicle (1% DMSO) or the complexes
tical grade. All buffer components were of biological grade and
for 12 h. For microscopy studies, the cells grown on slides were
used as received. Ruthenium(III) chloride hydrate (Alfa Aesar),
washed twice with PBS, stained with medium containing 1 μg/
bpy (Alfa Aesar), phen (Alfa Aesar), DIP (Alfa Aesar), cisplatin
mL AO for 15 min, and examined immediately by fluorescence
(Acros), MTT (Sigma), 3-MA (Sigma), CQ (Sigma), PI (Sigma),
microscopy (Axio Imager Z1, Carl Zeiss, Germany) using
490 nm band-pass blue excitation filters and a 515 nm long-pass
4PF6 (Alfa Aesar) were used without purification.
barrier filter according to a published protocol.70 For flow cyto-
2Cl2] 3 2H2O,76,77complex 4,78 and NAMI-A79
were prepared according to reported methods. The purity of
metry studies, cells were removed from 60 mm dishes (Corning)
synthesized compounds was analyzed via reversed-phase HPLC
with trypsin-EDTA and washed twice with PBS. After being
and was found to be g95% pure. Stock solutions of cisplatin
stained with AO for 15 min, the cells were collected in phenol
(5 mM in PBS) and NAMI-A (10 mM in PBS) were freshly
red-free growth medium. Green (510-530 nm) and red (650 nm)
prepared for every experiment. Stock solutions (20 mM) of 1-4
fluorescence emission from 10 000 cells illuminated with blue
were prepared in DMSO, which were proved to be stable for at
(488 nm) excitation light was analyzed on a flow cytometer
least 48 h at room temperature as monitored by UV-visible
(Becton Dickinson, Franklin Lakes, NJ) using CellQuest
Cellular Uptake. Flow Cytometry. HeLa cells in growth
Intracellular ROS Measurement. After treatment with Ru(II)
medium were seeded in 35 mm tissue culture dishes (Corning)
complexes for 6 h, the cells were incubated with 10 μM H2DCF-
and incubated at 37 °C under a 5% CO
DA (Sigma-Aldrich) for 20 min at 37 °C. The fluorescence
2 atmosphere until 70%
confluent. The culture medium was removed and replaced with
intensity of cells was measured by flow cytometry (Becton
medium (final DMSO concentration, 1% v/v) containing the
Dickinson, Franklin Lakes, NJ), confocal microscope (Leica
Ru(II) complexes at 20 μM. After incubation for 2 h, the
Microsystems, Wetzlar, Germany), and microplate analyzer
cell layer was trypsinized and washed twice with cold PBS
(Infinite M200, TECAN, Switzerland) with excitation set at
(phosphate buffered saline). The samples were raised in 500 μL
488 nm and emission at 530 nm. When necessary, Tiron (5 mM)
of cold PBS and analyzed by a FACSCalibur flow cytometer
and NAC (10 mM) were applied 1 h before Ru(II) treatment and
(Becton Dickinson & Co., Franklin Lakes, NJ) immediately.
kept in the medium during Ru(II) treatment until the cells were
The samples were collected in FL2 channel (excitation at 488 nm
and emission at 585 ( 21 nm), and the number of cells analyzed
Inhibition of Apoptosis and Autophagy. HeLa cells were pre-
for each sample was 10 000.30
incubated with 25 μM CQ, 5 mM 3-MA, or 50 μM z-VAD-
Live Cell Confocal Microscopy. HeLa cells were grown on
FMK for 1 h before the complexes were added. Percentage of
chamber slides to 70% confluence. Complex 3 (5 μM) was added
apoptosis was analyzed by flow cytometry, and cell viability was
to the culture medium (final DMSO concentration, 0.1% v/v)
determined by MTT as described in Supporting Information.
and incubated for varying amounts of time at 37 °C. The cells
Statistical Analysis. All biological experiments were per-
were then washed with PBS (2 � 200 μL) and photographed with
formed at least twice with triplicates in each experiment. Re-
a Leica TCS SP5 confocal microscope (Leica Microsystems,
presentative results were depicted in this report. Data were
Wetzlar, Germany) using a planapochromate 63�/NA 1.4 oil
presented as mean values ( standard deviations, and compar-
immersion objective. The confocal microscope was equipped
isons were made using Student's t test (two-tailed). A probabil-
with an ArKr laser which was used to excite Ru(II) (488 nm
ity of 0.01 or less was considered statistically significant.
excitation, 600-620 nm emission).
GF-AAS (Graphite Furnace Atomic Absorption Spectro-
Acknowledgment. We thank Prof. Z. P. Chen for providing
metry). HeLa cells were seeded in 60 mm tissue culture dishes
the GFP-LC3 expression vector. This work is supported by
Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21 7623
National Basic Research Program (973 Program, Grant
(17) Schatzschneider, U.; Niesel, J.; Ott, I.; Gust, R.; Alborzinia, H.;
2007CB815800), International S&T Cooperation Program
Wolfl, S. Cellular uptake, cytotoxicity, and metabolic profilingof human cancer cells treated with ruthenium(II) polypyridyl
(Grant 2007DFA30840), State High-Tech Development Pro-
complexes [Ru(bpy)2(N-N)]Cl2 with N-N = bpy, phen, dpq, dppz,
gram (863 Program, Grants 2006AA090504, 2007AA091401,
and dppn. ChemMedChem 2008, 3, 1104–1109.
and 2008AA09Z401), China Postdoctoral Science Founda-
(18) Cao, R. H.; Peng, W. L.; Wang, Z. H.; Xu, A. L. β-Carboline
tion (Grant 20080440790), Key Projects of Commission of
alkaloids: biochemical and pharmacological functions. Curr. Med.
Chem. 2007, 14, 479–500.
Science and Technology of Guangdong Province and
(19) Xiao, S. L.; Lin, W.; Wang, C.; Yang, M. Synthesis and biological
Guangzhou City, and Project of Sun Yet-sen University
evaluation of DNA targeting flexible side-chain substituted beta-
Science Foundation. A.X. is a recipient of the "Outstanding
carboline derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 437–441.
Young Scientist Award" from the National Natural Science
(20) Funayama, Y.; Nishio, K.; Wakabayashi, K.; Nagao, M.; Shimoi,
K.; Ohira, T.; Hasegawa, S.; Saijo, N. Effects of β- and γ-carboline
Foundation of China.
derivatives on DNA topoisomerase activities. Mutat. Res., Fun-dam. Mol. Mech. Mutagen. 1996, 349, 183–191.
(21) Li, Y.; Liang, F. S.; Jiang, W.; Yu, F. S.; Cao, R. H.; Ma, Q. H.;
Supporting Information Available: Details of methods; tables
Da, X. Y.; Jiang, J. D.; Wang, Y. C.; So, S. DH334, a β-carboline
of crystallographic data; ORTEP diagram; NMR, ESI-MS, and
anti-cancer drug, inhibits the CDK activity of budding yeast.
UV-vis spectra; effects on cell distribution; effects of autop-
Cancer Biol. Ther. 2007, 6, 1193–1199.
hagy inhibitors; and two crystallographic files in CIF format.
(22) Castro, A. C.; Dang, L. C.; Soucy, F.; Grenier, L.; Mazdiyasni, H.;
This material is available free of charge via the Internet at http://
Hottelet, M.; Parent, L.; Pien, C.; Palombella, V.; Adams, J. Novel
pubs.acs.org. X-ray crystallographic data for 1-Py-βC and
IKK inhibitors: β-carbolines. Bioorg. Med. Chem. Lett. 2003, 13,
complex 1 have been deposited in the Cambridge Crystallo-
(23) Al-Allaf, T. A. K.; Ayoub, M. T.; Rashan, L. J. Synthesis and
graphic Data Centre under the accession numbers CCDC
characterization of novel biologically-active platinum(II) and
771224 and 771225, respectively.
palladium(II) complexes of some β-carboline alkaloids. J. Inorg.
Biochem. 1990, 38, 47–56.
(24) Al-Allaf, T. A. K.; Rashan, L. J. Synthesis and cytotoxic evaluation
of the first trans-palladium(II) complex with naturally occurring
(1) Kroemer, G.; Levine, B. Autophagic cell death: the story of a
alkaloid harmine. Eur. J. Med. Chem. 1998, 33, 817–820.
misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010.
(25) Alford, P. C.; Cook, M. J.; Lewis, A. P.; Mcauliffe, G. S. G.;
(2) Hotchkiss, R. S.; Strasser, A.; McDunn, J. E.; Swanson, P. E. Cell
Skarda, V.; Thomson, A. J.; Glasper, J. L.; Robbins, D. J. Lumi-
death. N. Engl. J. Med. 2009, 361, 1570–1583.
nescent metal complexes. Part 5. Luminescence properties of ring-
(3) Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of auto-
substituted 1,10-phenanthroline tris-complexes of ruthenium(II).
phagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967.
J. Chem. Soc., Perkin Trans. 2 1985, 705–709.
(4) White, E.; DiPaola, R. S. The double-edged sword of autophagy
(26) Puckett, C. A.; Barton, J. K. Methods to explore cellular uptake of
modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316.
ruthenium complexes. J. Am. Chem. Soc. 2007, 129, 46–47.
(5) Bellodi, C.; Lidonnici, M. R.; Hamilton, A.; Helgason, G. V.;
(27) Rillema, D. P.; Jones, D. S.; Woods, C.; Levy, H. A. Comparison of
Soliera, A. R.; Ronchetti, M.; Galavotti, S.; Young, K. W.; Selmi,
the crystal-structures of tris heterocyclic ligand complexes of
T.; Yacobi, R.; Van Etten, R. A.; Donato, N.; Hunter, A.;
ruthenium(II). Inorg. Chem. 1992, 31, 2935–2938.
Dinsdale, D.; Tirro, E.; Vigneri, P.; Nicotera, P.; Dyer, M. J.;
(28) Quaranta, A.; Lachaud, F.; Herrero, C.; Guillot, R.; Charlot,
Holyoake, T.; Salomoni, P.; Calabretta, B. Targeting autophagy
M. F.; Leibl, W.; Aukauloo, A. Influence of the protonic state of
potentiates tyrosine kinase inhibitor-induced cell death in Phila-
an imidazole-containing ligand on the electrochemical and photo-
delphia chromosome-positive cells, including primary CML stem
physical properties of a ruthenium(II)-polypyridine-type complex.
cells. J. Clin. Invest. 2009, 119, 1109–1123.
Chem.—Eur. J. 2007, 13, 8201–8211.
(6) Moretti, L.; Yang, E. S.; Kim, K. W.; Lu, B. Autophagy signaling
(29) Ghezzi, A.; Aceto, M.; Cassino, C.; Gabano, E.; Osella, D. Uptake
in cancer and its potential as novel target to improve anticancer
of antitumor platinum(II)-complexes by cancer cells, assayed by
therapy. Drug Resist. Updates 2007, 10, 135–143.
inductively coupled plasma mass spectrometry (ICP-MS). J. Inorg.
(7) Rosenfeldt, M. T.; Ryan, K. M. The role of autophagy in tumour
Biochem. 2004, 98, 73–78.
development and cancer therapy. Expert Rev. Mol. Med. 2009, 11,
(30) Lo, K. K.; Lee, T. K.; Lau, J. S.; Poon, W. L.; Cheng, S. H.
Luminescent biological probes derived from ruthenium(II) estra-
(8) Bruijnincx, P. C.; Sadler, P. J. New trends for metal complexes with
diol polypyridine complexes. Inorg. Chem. 2008, 47, 200–208.
anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206.
(31) Puckett, C. A.; Barton, J. K. Mechanism of cellular uptake of a
(9) Vessieres, A.; Top, S.; Pigeon, P.; Hillard, E.; Boubeker, L.;
ruthenium polypyridyl complex. Biochemistry 2008, 47, 11711–
Spera, D.; Jaouen, G. Modification of the estrogenic properties
of diphenols by the incorporation of ferrocene. Generation of
(32) Noor, F.; Wustholz, A.; Kinscherf, R.; Metzler-Nolte, N. A
antiproliferative effects in vitro. J. Med. Chem. 2005, 48, 3937–
cobaltocenium-peptide bioconjugate shows enhanced cellular up-
take and directed nuclear delivery. Angew. Chem., Int. Ed. 2005, 44,
(10) Meggers, E. Targeting proteins with metal complexes. Chem.
Commun. 2009, 1001–1010.
(33) Puckett, C. A.; Ernst, R. J.; Barton, J. K. Exploring the cellular
(11) Smalley, K. S.; Contractor, R.; Haass, N. K.; Kulp, A. N.; Atilla-
accumulation of metal complexes. Dalton Trans. 2010, 39, 1159–
Gokcumen, G. E.; Williams, D. S.; Bregman, H.; Flaherty, K. T.;
Soengas, M. S.; Meggers, E.; Herlyn, M. An organometallic
(34) Yan, Y.; Su, X.; Liang, Y.; Zhang, J.; Shi, C.; Lu, Y.; Gu, L.; Fu, L.
protein kinase inhibitor pharmacologically activates p53 and
Emodin azide methyl anthraquinone derivative triggers mitochon-
induces apoptosis in human melanoma cells. Cancer Res. 2007,
drial-dependent cell apoptosis involving in caspase-8-mediated Bid
67, 209–217.
cleavage. Mol. Cancer Ther. 2008, 7, 1688–1697.
(12) Bergamo, A.; Sava, G. Ruthenium complexes can target determi-
(35) Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008,
nants of tumour malignancy. Dalton Trans. 2007, 1267–1272.
27, 6194–6206.
(13) Hartinger, C. G.; Jakupec, M. A.; Zorbas-Seifried, S.; Groessl, M.;
(36) Shao, Y.; Gao, Z.; Marks, P. A.; Jiang, X. Apoptotic and auto-
Egger, A.; Berger, W.; Zorbas, H.; Dyson, P. J.; Keppler, B. K.
phagic cell death induced by histone deacetylase inhibitors. Proc.
KP1019, a new redox-active anticancer agent-preclinical develop-
Natl. Acad. Sci. U.S.A. 2004, 101, 18030–18035.
ment and results of a clinical phase I study in tumor patients. Chem.
(37) Rubinsztein, D. C.; Gestwicki, J. E.; Murphy, L. O.; Klionsky,
Biodiversity 2008, 5, 2140–2155.
D. J. Potential therapeutic applications of autophagy. Nat. Rev.
(14) Peacock, A. F. A.; Sadler, P. J. Medicinal organometallic chem-
Drug Discovery 2007, 6, 304–312.
istry: designing metal arene complexes as anticancer agents.
(38) Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S. J.; Gibson, S. B.
Chem.—Asian J. 2008, 3, 1890–1899.
Oxidative stress induces autophagic cell death independent of
(15) Suss-Fink, G. Arene ruthenium complexes as anticancer agents.
apoptosis in transformed and cancer cells. Cell Death Differ.
Dalton Trans. 2010, 39, 1673–1688.
2008, 15, 171–182.
(16) Hotze, A. C. G.; Bacac, M.; Velders, A. H.; Jansen, B. A. J.;
(39) Kanzawa, T.; Germano, I. M.; Komata, T.; Ito, H.; Kondo, Y.;
Kooijman, H.; Spek, A. L.; Haasnoot, J. G.; Reedijk, J. New
Kondo, S. Role of autophagy in temozolomide-induced cyto-
cytotoxic and water-soluble bis(2-phenylazopyridine)ruthenium-
toxicity for malignant glioma cells. Cell Death Differ. 2004, 11,
(II) complexes. J. Med. Chem. 2003, 46, 1743–1750.
7624 Journal of Medicinal Chemistry, 2010, Vol. 53, No. 21
(40) Seglen, P. O.; Gordon, P. B. 3-Methyladenine: specific inhibitor of
(62) Eisenberg-Lerner, A.; Bialik, S.; Simon, H. U.; Kimchi, A. Life and
autophagic/lysosomal protein degradation in isolated rat hepato-
death partners: apoptosis, autophagy and the cross-talk between
cytes. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 1889–1892.
them. Cell Death Differ. 2009, 16, 966–975.
(41) Maclean, K. H.; Dorsey, F. C.; Cleveland, J. L.; Kastan, M. B.
(63) Dalby, K. N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B.
Targeting lysosomal degradation induces p53-dependent cell death
Targeting the prodeath and prosurvival functions of auto
and prevents cancer in mouse models of lymphomagenesis. J. Clin.
phagy as novel therapeutic strategies in cancer. Autophagy 2010,
Invest. 2008, 118, 79–88.
6, 322–329.
(42) Wang, C. X.; Youle, R. J. The role of mitochondria in apoptosis.
(64) Sy, L. K.; Yan, S. C.; Lok, C. N.; Man, R. Y.; Che, C. M.
Annu. Rev. Genet. 2009, 43, 95–118.
Timosaponin A-III induces autophagy preceding mitochondria-
(43) Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J. J. Selective degra-
mediated apoptosis in HeLa cancer cells. Cancer Res. 2008, 68,
dation of mitochondria by mitophagy. Arch. Biochem. Biophys.
2007, 462, 245–253.
(65) Herman-Antosiewicz,
(44) Nowak, G. Protein kinase C-R and ERK1/2 mediate mitochondrial
Sulforaphane causes autophagy to inhibit release of cytochrome
dysfunction, decreases in active Naþ transport, and cisplatin-
C and apoptosis in human prostate cancer cells. Cancer Res. 2006,
induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–
66, 5828–5835.
(66) Qadir, M. A.; Kwok, B.; Dragowska, W. H.; To, K. H.; Le, D.;
(45) Ozben, T. Oxidative stress and apoptosis: impact on cancer ther-
Bally, M. B.; Gorski, S. M. Macroautophagy inhibition sensitizes
apy. J. Pharm. Sci. 2007, 96, 2181–2196.
tamoxifen-resistant breast cancer cells and enhances mitochondrial
(46) Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regula-
depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403.
tion of autophagy. Trends Cell Biol. 2007, 17, 422–427.
(67) Harhaji-Trajkovic, L.; Vilimanovich, U.; Kravic-Stevovic, T.;
(47) Kaufmann, S. H.; Hengartner, M. O. Programmed cell death: alive
Bumbasirevic, V.; Trajkovic, V. AMPK-mediated autophagy in-
and well in the new millennium. Trends Cell Biol. 2001, 11, 526–534.
hibits apoptosis in cisplatin-treated tumour cells. J. Cell. Mol. Med.
(48) Kang, K. B.; Zhu, C.; Yong, S. K.; Gao, Q.; Wong, M. C.
2009, 13, 3644–3654.
Enhanced sensitivity of celecoxib in human glioblastoma cells:
(68) Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.;
induction of DNA damage leading to p53-dependent G1 cell cycle
Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer
arrest and autophagy. Mol. Cancer 2009, 8, 66.
cells to radiation involves autophagy and formation of acidic
(49) Abedin, M. J.; Wang, D.; McDonnell, M. A.; Lehmann, U.;
vesicles. Cancer Res. 2001, 61, 439–444.
Kelekar, A. Autophagy delays apoptotic death in breast cancer
(69) Zhou, W. J.; Deng, R.; Zhang, X. Y.; Feng, G. K.; Gu, L. Q.;
cells following DNA damage. Cell Death Differ. 2007, 14, 500–510.
Zhu, X. F. G-quadruplex ligand SYUIQ-5 induces autophagy by
(50) Nair, R. B.; Teng, E. S.; Kirkland, S. L.; Murphy, C. J. Synthesis
telomere damage and TRF2 delocalization in cancer cells. Mol.
and DNA-binding properties of [Ru(NH3)4dppz]2. Inorg. Chem.
Cancer Ther. 2009, 8, 3203–3213.
1998, 37, 139–141.
(70) Kanzawa, T.; Kondo, Y.; Ito, H.; Kondo, S.; Germano, I. Induc-
(51) Haq, I.; Lincoln, P.; Suh, D.; Norden, B.; Chowdhry, B. Z.;
tion of autophagic cell death in malignant glioma cells by arsenic
Chaires, J. B. Interaction of Δ- and Λ-[Ru(phen)2DPPZ]2þ with
trioxide. Cancer Res. 2003, 63, 2103–2108.
DNA: a calorimetric and equilibrium binding study. J. Am. Chem.
(71) Kanzawa, T.; Zhang, L.; Xiao, L. C.; Germano, I. M.; Kondo, Y.;
Soc. 1995, 117, 4788–4796.
Kondo, S. Arsenic trioxide induces autophagic cell death in
(52) Chen, Q.; Chao, R.; Chen, H.; Hou, X.; Yan, H.; Zhou, S.;
malignant glioma cells by upregulation of mitochondrial cell death
Peng, W.; Xu, A. Antitumor and neurotoxic effects of novel
protein BNIP3. Oncogene 2005, 24, 980–991.
harmine derivatives and structure-activity relationship analysis.
(72) Opipari, A. W., Jr.; Tan, L.; Boitano, A. E.; Sorenson, D. R.;
Int. J. Cancer 2005, 114, 675–682.
Aurora, A.; Liu, J. R. Resveratrol-induced autophagocytosis in
(53) Timerbaev, A. R.; Hartinger, C. G.; Aleksenko, S. S.; Keppler,
ovarian cancer cells. Cancer Res. 2004, 64, 696–703.
B. K. Interactions of antitumor metallodrugs with serum proteins:
(73) Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J. P.;
advances in characterization using modern analytical methodol-
Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell
ogy. Chem. Rev. 2006, 106, 2224–2248.
death in chronic myelogenous leukemia cells via JNK-mediated
(54) Meggers, E. Exploring biologically relevant chemical space with
p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010,
metal complexes. Curr. Opin. Chem. Biol. 2007, 11, 287–292.
70, 1042–1052.
(55) Maksimoska, J.; Feng, L.; Harms, K.; Yi, C.; Kissil, J.; Marmorstein,
(74) Salazar, M.; Carracedo, A.; Salanueva, I. J.; Hernandez-Tiedra, S.;
R.; Meggers, E. Targeting large kinase active site with rigid, bulky
Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.;
octahedral ruthenium complexes. J. Am. Chem. Soc. 2008, 130,
Garcia, S.; Nowak, J.; Fimia, G. M.; Piacentini, M.; Cecconi, F.;
Pandolfi, P. P.; Gonzalez-Feria, L.; Iovanna, J. L.; Guzman, M.;
(56) Neugebauer, U.; Pellegrin, Y.; Devocelle, M.; Forster, R. J.;
Boya, P.; Velasco, G. Cannabinoid action induces autophagy-
Signac, W.; Moran, N.; Keyes, T. E. Ruthenium polypyridyl
mediated cell death through stimulation of ER stress in human
peptide conjugates: membrane permeable probes for cellular imag-
glioma cells. J. Clin. Invest. 2009, 119, 1359–1372.
ing. Chem. Commun. 2008, 5307–5309.
(75) Guenther, G. G.; Peralta, E. R.; Rosales, K. R.; Wong, S. Y.;
(57) Puckett, C. A.; Barton, J. K. Fluorescein redirects a ruthenium-
Siskind, L. J.; Edinger, A. L. Ceramide starves cells to death by
octaarginine conjugate to the nucleus. J. Am. Chem. Soc. 2009, 131,
downregulating nutrient transporter proteins. Proc. Natl. Acad.
Sci. U.S.A. 2008, 105, 17402–17407.
(58) O'Connor, N. A.; Stevens, N.; Samaroo, D.; Solomon, M. R.;
(76) Sullivan, B.; Salmon, D.; Meyer, T. Mixed phosphine 2,20-bipyr-
Marti, A. A.; Dyer, J.; Vishwasrao, H.; Akins, D. L.; Kandel, E. R.;
idine complexes of ruthenium. Inorg. Chem. 1978, 17, 3334–3341.
Turro, N. J. A covalently linked phenanthridine-ruthenium(II)
(77) Caspar, R.; Cordier, C.; Waern, J. B.; Guyard-Duhayon, C.;
complex as a RNA probe. Chem. Commun. 2009, 2640–2642.
Gruselle, M.; Le Floch, P.; Amouri, H. A new family of mono-
(59) Musatkina, E.; Amouri, H.; Lamoureux, M.; Chepurnykh, T.;
and dicarboxylic ruthenium complexes [Ru(DIP)2)(L2)]2þ (DIP =
Cordier, C. Mono- and dicarboxylic polypyridyl-Ru complexes as
4,7-diphenyl-1,10-phenanthroline): synthesis, solution behavior,
potential cell DNA dyes and transfection agents. J. Inorg. Biochem.
and X-ray molecular structure of trans-[Ru(DIP)2(MeOH)2]-
2007, 101, 1086–1089.
[OTf]2. Inorg. Chem. 2006, 45, 4071–4078.
(60) Tu, L. C.; Chen, C. S.; Hsiao, I. C.; Chern, J. W.; Lin, C. H.; Shen,
(78) Hartshorn, R. M.; Barton, J. K. Novel dipyridophenazine com-
Y. C.; Yeh, S. F. The β-carboline analog Mana-Hox causes mitotic
plexes of ruthenium(II): exploring luminescent reporters of DNA.
aberration by interacting with DNA. Chem. Biol. 2005, 12, 1317–1324.
J. Am. Chem. Soc. 1992, 114, 5919–5925.
(61) Pommier, Y.; Sordet, O.; Antony, S.; Hayward, R. L.; Kohn, K. W.
(79) Mestroni, G.; Alessio, E.; Sava, G. New Salts of Anionic Com-
Apoptosis defects and chemotherapy resistance: molecular inter-
plexes of Ru(III), as Antimetastatic and Antineoplastic Agents.
action maps and networks. Oncogene 2004, 23, 2934–2949.
PCT Int. Appl. WO 98/00431, 1998.
Source: http://home.sysu.edu.cn/yysh/refsys/uppdf/20118120322773499.pdf
LIECHTENSTEINER VATERLAND DONNERSTAG, 3. MAI 2012 29 Ein aktiver Wanderer kommt zurück erwartetNew York. – Eines der berühmtes- Sie sind zurück – die pelzigen ten Gemälde der modernen Kunst und fleissigen Nager. Am Mitt- kam in der zurückliegenden Nacht wochabend führte Holger Frick,
Curriculum Vitae JACK MERRIT GWALTNEY, JR. December 24, 1930, Norfolk, Virginia B.A. University of Virginia 1948-1952 M.D. University of Virginia 1952-1956 Summary of Career: University Hospitals of Cleveland, Cleveland, Ohio Residency, Internal Medicine University Hospitals of Cleveland, Cleveland, Ohio Chief Resident, Internal University of Virginia Hospital


