
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
The interplay of structure and dynamics: insights from a survey of hiv1 reverse transcriptase crystal structures
STRUCTURE O FUNCTION O BIOINFORMATICS
The interplay of structure and dynamics:Insights from a survey of HIV-1 reversetranscriptase crystal structures
James M. Seckler,1 Nicholas Leioatts,2 Hongyu Miao,1 and Alan Grossfield2*1 Department of Biostatistics and Computational Biology, University of Rochester, Rochester, New York2 Department of Biochemistry and Biophysics, University of Rochester, Rochester, New York
HIV-1 reverse transcriptase (RT) is a critical drug target for HIV treatment, and understanding the exact mechanisms of itsfunction and inhibition would significantly accelerate the development of new anti-HIV drugs. It is well known that struc-ture plays a critical role in protein function, but for RT, structural information has proven to be insufficient—despite enor-mous effort—to explain the mechanism of inhibition and drug resistance of non-nucleoside RT inhibitors. We hypothesizethat the missing link is dynamics, information about the motions of the system. However, many of the techniques that givethe best information about dynamics, such as solution nuclear magnetic resonance and molecular dynamics simulations,cannot be easily applied to a protein as large as RT. As an alternative, we combine elastic network modeling with simultane-ous hierarchical clustering of structural and dynamic data. We present an extensive survey of the dynamics of RT bound toa variety of ligands and with a number of mutations, revealing a novel mechanism for drug resistance to non-nucleoside RTinhibitors. Hydrophobic core mutations restore active-state motion to multiple functionally significant regions of HIV-1 RT.
This model arises out of a combination of structural and dynamic information, rather than exclusively from one or theother.
Proteins 2013; 81:1792–1801.
C 2013 Wiley Periodicals, Inc.
Key words: elastic network model; allostery; reverse transcriptase inhibition; protein–drug interactions; structure–function.
There are three classes of inhibitors to RT: nucleoside/
nucleotide reverse transcriptase inhibitors (NRTI/NtRTI),
HIV-1 reverse transcriptase (RT) has long been a
major target for anti-HIV therapies. Understanding its
(NNRTIs), and RNase H inhibitors (RIs). NRTIs are con-
function and inhibition is important for designing new
verted to nucleotide analogs in the body, but lack a 30-
inhibitors. Moreover, there is a huge amount of struc-
OH, which allows them to act as chain terminators.5
tural information available: there are over 100 crystal
NtRTIs behave in the same fashion as NRTIs but do not
structures, including native and mutant proteins with
require the conversion step in the body. NNRTIs are
various ligands bound. RT is a multifunctional enzyme
small molecules that bind to a pocket inside the palm
that turns single-strand viral RNA into double-stranded
subdomain of p66 and allosterically inhibit all polymer-
DNA, giving it a crucial role in viral infectivity. The
ase activity and polymerase-dependent RNase H activity;
structure of RT is a heterodimer with a larger 66 kDa
surprisingly, they accelerate polymerase-dependent RNase
subunit (p66), consisting of a polymerase domain, whichin turn contains several subdomains: the fingers, palm,thumb, and connection subdomains, as well as an RNase
Additional Supporting Information may be found in the online version of thisarticle.
H domain (Fig. 1). The smaller 51 kDa subunit (p51)
has the same N-terminal sequence as p66 but lacks the
HHSN272201000055C, P30 AI078498, GM068411, and GM095496.
*Correspondence to: Alan Grossfield, Department of Biochemistry and Biophy-
RNase H domain.1 The p66 subunit is thought to con-
sics, University of Rochester Medical Center, 601 Elmwood Ave, Box 712, Roches-
tain all of the functionally important features of RT,
ter, NY 14642. E-mail:
[email protected]
whereas p51 is thought to provide stability and aid in
Received 29 November 2012; Revised 12 April 2013; Accepted 19 April 2013Published online 30 May 2013 in Wiley Online Library (wileyonlinelibrary.com).
allosteric communication across the protein.1–4
DOI: 10.1002/prot.24325
C 2013 WILEY PERIODICALS, INC.
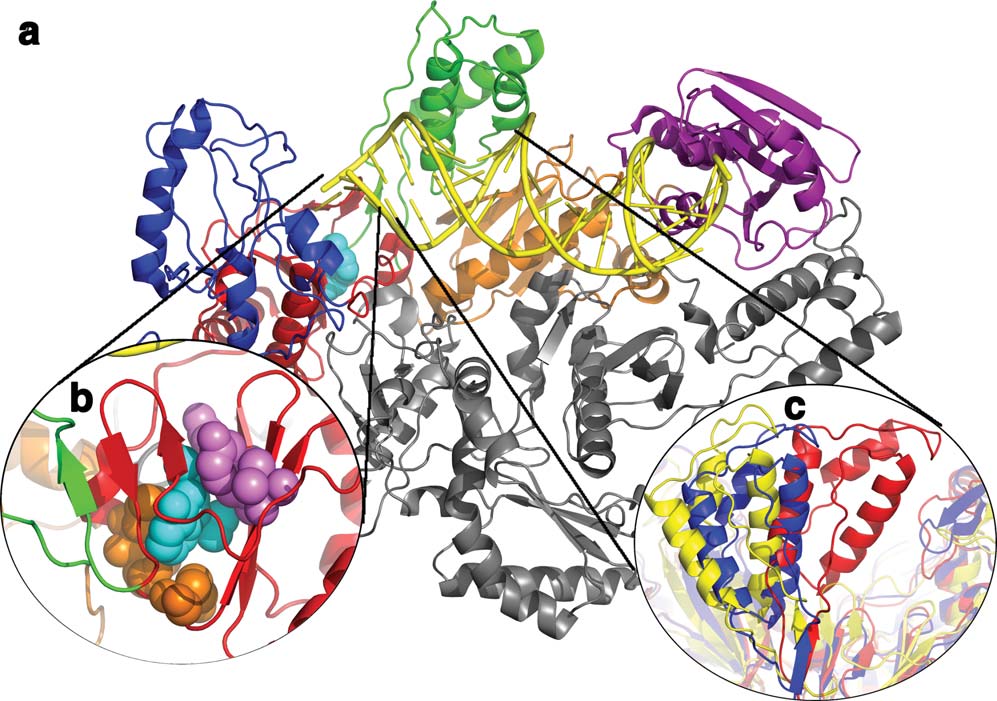
Structure and Dynamics of HIV RT
The structure of HIV-1 RT containing the larger subunit (p66) has a polymerase domain consisting of a fingers (blue), palm (red), thumb (green),and connection (orange) subdomain and an RNase H (purple) domain. The smaller subunit (p51) has the same N-terminal sequence as p66(gray), but lacks the RNase H domain. (b) The NNRTI binding pocket with the NNRTI (cyan, spheres) and drug resistant mutants shown inspheres colored by if they are hydrophobic core mutations (purple) or entry blocker mutations (orange). (c) The change in the position of thethumb subdomain depending on which ligand RT is bound to: unliganded (red; 1DLO), DNA bound (blue; 1N5Y), or NNRTI bound (yellow;1VRT).54
H activity.6–8 They function by preventing the DNA-
wild type and hydrophobic core mutants is a subtle
bound protein from forming an active complex with
rotation of b-9 and b-11 with respect to the other b-
deoxyribonucleotide triphosphate (dNTP) to continue
sheet that makes up the drug binding pocket (b-12-13-
chain elongation.9 NNRTIs are divided into three gener-
14).16–20 The exact mechanism of these allosteric
ations, with each generation better able to form stable
NNRTI-resistance mutations is particularly mysterious.
hydrogen bonds and hydrophobic interactions with the
Exploring the mechanism of RT inhibition and drug
drug binding pocket.10 RIs are the newest class of RT
resistance has spawned a wealth of crystallographic infor-
inhibitors, small molecules that bind 50 A˚ away from the
mation. Recently, there have been many attempts to use
RNase H active site, near the polymerase active site.11
clustering or other methods to survey this crystallo-
There are currently 16 RT inhibitors approved for clinical
graphic data, focusing either on the shape of the NNRTI,
use, including nine NRTIs, two NtRTIs, and six NNRTIs.
the binding pocket residues, or B-factors.21–23 All of
At this time, there are no clinically approved RIs.12,13
There are thought to be three types of NNRTI resist-
between RT bound to various ligands, but to date there
ance mutations: entrance, deformation, and hydrophobic
is no method that can correctly predict the functional
core mutations. Entrance mutations (K103N and K101E)
state of the protein (e.g., inhibited, active, etc.) based
are thought to block drug entry into the binding
solely on the crystal structure. Surveying a large number
pocket.14,15 Deformation mutations (L100I and G190S)
of crystal structures and determining meaningful infor-
change the shape of the drug binding pocket, making
mation from them, particularly in a quantitative way,
binding unfavorable.16,17 Hydrophobic core mutations
remains a major challenge. This arises from the fact that
(V108I, Y181C, and Y188C) interrupt ring stacking inter-
each crystal structure contains an enormous amount of
actions with the drug, conveying resistance, presumably
information, but paradoxically, structural data alone is
by reducing the binding affinity by eliminating hydro-
not always sufficient to determine a protein's function.
phobic interactions between the NNRTI and the hydro-
Thus, figuring out precisely which differences between
phobic core of the binding pocket. The primary
closely related structures are important (and why)
difference between structures with an NNRTI bound to
remains an unsolved problem. Given the challenges
J.M. Seckler et al.
inherent in exploring these issues experimentally, compu-
require that the various structural models contain an
tational approaches are extremely attractive. The obvious
identical number of atoms. We then removed extraneous
first choice would be to use all-atom molecular dynam-
regions from the remaining structures leaving a consen-
ics, because this is the gold standard for biomolecular
simulation. Unfortunately, these kinds of calculations are
Table S1 shows the sequences of all structures used in
very expensive to perform, and for a system as large as
this study, where "-" represents missing sequence. How-
RT would likely require multiple microseconds of sam-
ever, regions of removed sequence were taken into
pling to achieve even a semblance of statistical conver-
account by using vibrational subset analysis (VSA)34 (see
gence.24,25 In the present context, where we wish to
the following section for additional details).
tease out subtle differences between a large number of
Of the original 54 X-ray structures considered, two
similar structures, these statistical errors would almost
(3HVT and 1LWC) were excluded due to excessive miss-
certainly swamp out the desired signal. As a result, we
ing sequence coverage. The remaining 52 structures were
instead turn to more approximate (and thus less expen-
aligned and residues were removed until all 52 structures
sive) modeling techniques.
had the same set of missing sequence (Supporting Infor-
Elastic network modeling, particularly using the aniso-
mation Table S1). The side chains of the remaining resi-
tropic network model (ANM), is a powerful tool for
dues were removed, and all calculations were performed
quickly probing the local protein energy landscape and
using the Ca atoms. ANM was then performed on all 52
extracting the coherent motions available to the sys-
structures using VSA to model in all removed sequences,
tem.26,27 This model works particularly well on systems
and the resulting eigenvalues and eigenvectors were
that are too large to be characterized by all-atom molec-
saved. This resulted in a final data set containing two
ular dynamics, allowing the investigation of the mecha-
strains of HIV-RT, spanning multiple crystallographic
nistic properties of the protein, the location of active
space groups (Supporting Information Table S2).26,34
sites, and allosteric causes of drug resistance. ANMs havebeen applied to proteins such as HIV-1 protease, as wellas complex and large systems such as the entire microtu-
Anisotropic network modeling
bule complex.28–31 Furthermore, we previously showed
An ANM represents the protein as a network of beads
that the motions predicted by ANMs compare well with
connected by springs, typically each bead representing
long molecular dynamics trajectories, despite the simpli-
the position of a Ca. The potential energy between the
fying assumptions built into the methodology.25,32 By
ith and jth Ca in the network is given by Hooke's law:
surveying both the structure and the dynamics of a setof proteins, we are able to elucidate functionally impor-
tant structural changes.
Here, we report that NNRTI binding shifts both the
structure and dynamics of RT, and that hydrophobic
ðxo2xoÞ21ðyo2yoÞ21ðzo2zoÞ2 is the dis-
core mutations restore the motions of the active sites
tance between atoms i and j in the reference structure,
and dNTP binding site to those of the uninhibited struc-
and Cij is the spring constant.25,26 The reference struc-
ture. Apparently similar protein structures can have very
ture is by definition the minimum energy structure,
different dynamic fingerprints, so clustering by both
because vij � 0, and is only 0 at d 5 do. The spring con-
structure and dynamics is uniquely valuable for under-
stant is defined as Cij 5 1 within a cutoff distance of
standing protein function.
15 A˚ and 0 beyond it. Using this connection rule, a Hes-sian matrix of the potential is constructed. This yields a3N 3 3N matrix, where N is the number of nodes in the
network. When diagonalized, this matrix returns eigen-values (k
X-ray structure selection and analysis
i) and eigenvectors ð
miÞ corresponding to the
vibrational modes of the protein. The eigenvectors are
Crystallographic data were obtained from the Protein
the directions of motion, with each associated eigenvalue
Data Bank.33 All unliganded, DNA, RNA, and adenosine
triphosphate (ATP) bound X-ray structures were initially
Because the protein is modeled as a harmonic system,
selected, along with all X-ray structures of HIV-1 RT
bound to the first generation NNRTI Nevirapine, the
amplitude of motion, meaning that the largest scale
second generation NNRTI Efavirenz, and the third gener-
motions will be those with the lowest frequencies. The
ation NNRTIs etravirine, rilpivirine, and lersivirine. We
six lowest frequency modes, corresponding to rigid body
aligned the sequences and structures of these molecules,
translation and rotation, are ignored for all subsequent
and identified regions that were absent in some struc-
tures. We the excluded those X-ray structures with signif-
Not all structures have the same atoms resolved, but
icant regions of unresolved structure because our analysis
the results of the eigendecompositions can only be
Structure and Dynamics of HIV RT
mA are the ith eigenvalue and eigenvector
of structure A, and kB and
mB are the jth eigenvalue and
eigenvector of structure B. The covariance complement is0 when the two ANM eigeinsets are the same, and 1when they are completely orthogonal. In contrast toother methods for comparing results of ANMs, such asthe subset overlap, the covariance overlap and covariancecomplement directly take the eigenvalue spectrum—therelative importance of specific mode—into account.
Agglomerative hierarchical clustering using average
linkage was used to classify X-ray structures by the ratioof their root-mean-square deviation (RMSD) to covari-
ance complement.36 This takes advantage of the fact that
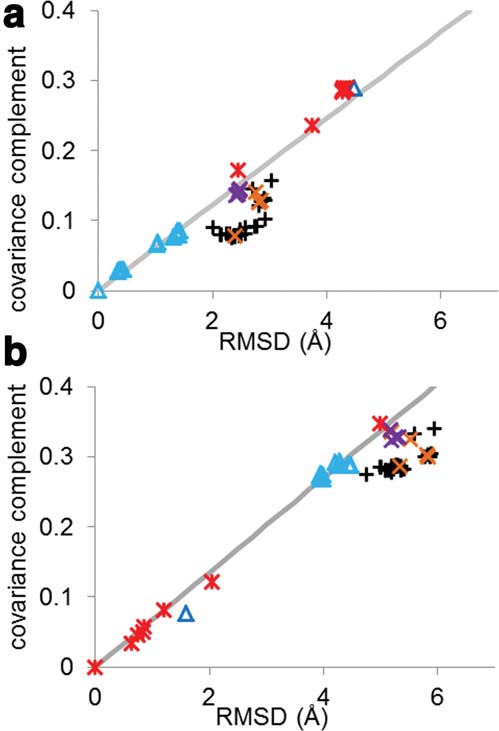
The covariance complement and RMSD of all 52 structures compared
structures with like functions to the reference structure
to (a) wild-type RT bound to DNA (1N6Q) and (b) unliganded RT(1DLO). Points are shaped and colored by the ligand and mutations:
show a linear relationship between RMSD and covariance
NNRTI (black pluses), RT bound to DNA (light blue triangles), RNA
complement with the line having a y-intercept of 0
(dark blue triangle), unliganded (red stars), entry blocker mutants
(Fig. 2). Three clusters were formed corresponding to an
bound to susceptible NNRTI (orange Xs), and hydrophobic core
active, preactive, and inactive states.
mutants bound to susceptible NNRTI (purple Xs). The best fit line toeither all DNA-bound RT (a) or unliganded RT (b) is shown in gray.
Structures that show a linear relationship between RMSD and covari-
Covariance matrices
ance complement tend to show similar functional abilities, whereas pro-teins that form off diagonal clusters tend to have different functional
To probe differences in motion within and between
abilities. This is true even for very different structures (DNA bound
clusters of proteins, we computed the inter-residue
and unliganded).54,55
covariance matrix for the first 50 modes of allproteins37:
readily compared when the matrix dimensions are identi-cal. To avoid excluding available information, we applied
a recently developed variation of ANMs, VSA to account
for the extra residues. VSA partitions the Hessian matrix
into an environment and a subsystem.34 Here, the sub-
system is our consensus residues, which are diagonalized
as in ANM. The environment is comprised of the extra
residues, which get diagonalized separately. The fluctua-
i,j is the covariance between the ith and jth res-
tions of the environment are integrated out, leaving only
l,i and kl are the eigenvectors and eigenvalue of
the lth mode. This matrix tracks the degree to which the
the environment's effect on the subsystem. This allows us
motions of various portions of the protein are related; a
to analyze the vibrational modes of a subset of amino
value of 1 indicates that the two residues move as a rigid
acids in p66, and model in the parts of the sequence that
body, 0 means they are independent, and 21 indicates
are not common to all structures.
anticorrelated movement. Because we surveyed 52 pro-teins in total, we need to further condense the data forinterpretation. Accordingly, we calculated the matrix of
Covariance complement
standard deviations of all CMs in a cluster (or between
We compared the ANM profiles of various p66
clusters); this reveals regions of the protein where motion
conformations to each other using a modified version
differs either within that cluster, or between cluster, and

J.M. Seckler et al.
are uniquely determined by the structure, it is not sur-prising that the two quantities show significant correla-tion. In Figure 2, we plotted correlation coefficientversus RMSD for unliganded [1DLO, Fig. 2 (a)] andDNA bound [1N6Q, Fig. 2(b)] RT. Structures capable ofthe same function tend to show a linear relationshipbetween covariance complement and RMSD, whereasstructures that are not capable of the same function arefound off the line. Surprisingly, this rule holds evenwhen the structures differ significantly. For example, theRMSD between 1DLO and 1RTJ (both unliganded RTstructures) is 5.00 A˚, but when all of the unligandedstructures are plotted, they can be fit linearly withR2 5 .9934 [Fig. 2(a), black line]. All wild-type RT struc-tures bound to NNRTIs deviate significantly from a lin-
ear relationship; this holds true regardless whether an
Agglomerative hierarchical clustering was used on the ratio of covari-
unliganded structure or a DNA-bound RT structure is
ance complement to RMSD, forming an active, preactive, and inactivecluster. The resulting clusters are colored by their ligand: NNRTI-
used as reference. All structures with a function similar
bound drug inhibited mutants (black), first/second Generation NNRTI-
to the reference structure fall on a line with a y-intercept
bound hydrophobic core mutants (purple), first/second Generation
NNRTI-bound entry blocker mutants (orange), DNA (light blue), RNA(dark blue), and unliganded (red). The sole structure bound to bothDNA and NNRTIs is striped black and light blue.
gives a value of 0 where the inter-residue motion is
Because linear variation of RMSD with covariance
unchanged. We then applied a Fisher transformation to
complement is a signature of functional commonality,
the covariance matrix:
we focus on the ratio of the two quantities. Specifically,we computed the ratio of RMSD to covariance comple-
ment for all pairs of structures, and performed agglomer-
ative hierarchical clustering on the resulting matrix. The
procedure produced three main clusters of structures,
to account for the fact that covariances are not normally
which we label "active," "inactive," and "preactive." The
clustering is shown in Figure 3 and Table I lists all 52 RT
unbounded, we assigned covariance matrix values greater
structures and which cluster they fall into. The active
60.99 a z-score of 62.5. We then performed a Welches t
cluster contains all RT structures bound to DNA except
test on every element of the resulting matrix of z-scores,
for 3V81, which is bound to both DNA and an NNRTI.
and compared the resulting patterns for variations within
The inactive cluster contains all structures where there is
and between clusters.39
an NNRTI bound to an RT it can inhibit. In addition,this cluster contains all of the structures of proteins withentry blocker drug resistance mutations bound to either
Computational analysis
first or second generation NNRTIs. The preactive cluster
ANM and covariance complement calculations were
contains all unliganded RT structures and hydrophobic
performed using the LOOS software package.40 All clus-tering was performed using Cluster 3.0.36 Computations
were performed on the University of Rochester research
linux cluster.
1R0A, 1N6Q, 1T03, 1N5Y, 3KLH, 1T05,
3JSM, 2HMI, 1J5O, 3KLG, 3KLE,
1S1X, 1JKH, 1LWF, 1JLF, 1FKP, 1JLB,
Structural comparisons
1RTJ, 1HVU, 1QE1, 2IAJ, 1HMV,
3KLI, 1DLO, 1HQE, 3DLK
The covariance complement [Eq. (2)] quantifies the
1FKO, 1LWE, 1S1U, 1LW0, 2HND,
similarity between the motions predicted for two struc-
2HNY, 1FK9, 1VRT, 3V81, 2WON,
tures, whereas RMSD quantifies their structural similar-
2WOM, 3LP0, 3LP1, 1IKW, 1IKV,
ity. The covariance complement and RMSD were
3M8P, 3MED, 3MEC, 3MEE, 3MEG,
3QIP, 1SV5, 2ZE2, 2ZD1, 3BGR
calculated between each X-ray structure in the set. Giventhat in an elastic network model (ENM), the dynamics
EFZ, efavirenz; NPV, nevirapine; ERT, etravirine; LVR, lersivirine; RIP, rilpivirine.
Structure and Dynamics of HIV RT
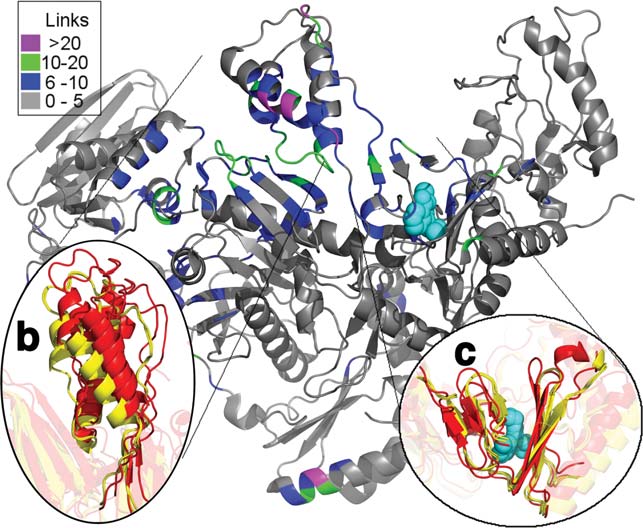
thumb subdomain rests, and the subtle rotation leads toa marked change in the positioning of the thumb subdo-main, shifting it away from the connection subdomain inthe NNRTI-bound drug resistant mutants [Fig. 4(b)].
This rigid-body motion of the thumb subdomain signifi-cantly changes interdomain contacts, resulting in hetero-geneity in the predicted dynamics. Figure 4(a) shows allresidues that form 5 or more additional contacts instructures in the inactive cluster versus the preactivestructures. Unsurprisingly, most of these residues fallalong the interface between the thumb and connectionsubdomains of p66. Moreover, all of these residues arepart of an experimentally determined network of alloste-ric tightening.4 This shows how a subtle change in thepositioning of a single subdomain can have radicaleffects on the predicted dynamics.
The structure of RT colored by the difference in contacts in the connec-tivity matrix (Cij) between the preactive to the inactive cluster. (b)
accounting for the difference in dynamics and function
shows the shift in the thumbs position between two structures in the
between the clusters, we next sought to identify specific
NNRTI-bound preactive cluster (red) and two structures from the
residues whose dynamics change between functional
NNRTI inactive cluster (yellow). The thumb subdomain rotates away
states. To do so, we looked at the motions predicted for
from the connection subdomain. (c) shows the subtle rotation in b-12-13-14 which forms half of the drug binding pocket.20,56–58
each residue, in the form of the inter-residue covariancemap (see Methods section); in short, these maps describe
core mutants bound to either first or second generation
the degree to which two residues' motions are related to
NNRTIs, as well as RNA-bound RT, and first generation
each other. These maps were computed for each struc-
NNRTI-bound RT with the K103N mutation, a particu-
ture, and were used to compare the variation within a
larly potent entry blocker mutation.12,41
cluster to the variation between clusters. As discussed inthe Methods, we identified the specific residues (or setsof residues) whose behavior differs significantly between
Intercluster structural differences
clusters. This method allows us to identify regions where
We first compared the structural variations between
the overall nature of the motion changes, for instance
the structures in the three clusters, to see if there is a
from correlated motion (blue on the graph, indicating
simple explanation for their classification. The structures
rigid body motion) to uncorrelated (white) or anticorre-
in the active cluster are very similar, with an RMSD of
lated (red, where the regions move in opposing
1.68 A˚ between the two least similar structures. The inac-
tive cluster is also fairly self-similar, with a maximum
In the preactive cluster, the fingers and palm subdo-
RMSD of 3.20 A˚. By contrast, the preactive cluster is far
main movements are correlated with those of the thumb
more diverse with a maximum RMSD of 5.63 A˚. The
subdomain and RNase H domain. By contrast, in the
variations within each cluster are not evenly distributed
inactive cluster the fingers and palm subdomain moves
throughout the structure; rather, the change primarily
as a rigid unit, and the thumb subdomain motions are
comes in the positioning of the thumb subdomain. The
correlated with the connection subdomain and the RNase
preactive cluster in particular shows the thumb subdo-
H domain (Fig. 5).
main in two major conformations [Fig. 1(c)], an unli-
The covariance maps for the preactive and active clus-
ganded position and a NNRTI-bound position; if one
ters are largely similar. In the active cluster, the thumb
breaks the preactive cluster into subclusters based on
subdomain is weakly correlated with the connection sub-
thumb position the maximum RMSD drops to 1.55 A˚
domain and RNase H domain, but otherwise shows a
for the NNRTI-bound thumb position and 2.08 A˚ for
similar inter-residue correlation. Inhibition radically
the Unliganded thumb position.
changes the predicted dynamics compared to both the
A common feature of all preactive structures is a rota-
active and preactive clusters: compared to active cluster,
tion of the b-sheet consisting of strand 12, 13, and 14
the inactive polymerase domain moves in a rigidly corre-
(b-12-13-14) relative to the inactive cluster's structures
lated manner (Supporting Information Fig. S1).
[Fig. 4(c)]. This rotation is subtle in the drug resistance
The hydrophobic core mutants in the preactive cluster
mutants, but this sheet is the platform upon which the
and the structures in the inactive clusters are structurally
J.M. Seckler et al.
in its molecular motions.3,44–46 Our results suggest thatsimple flexibility is not the sole determinant for proteinfunction; rather, it is the subtle interplay of structure andspecific motions that control function. This is shown bythe marked structural similarity of dynamics in the fin-gers and palm subdomains of the hydrophobic coremutants and the unliganded structures. It is also seen inthe similarities between the dynamics of the thumb andRNase H domain of the hydrophobic core mutants andthe active cluster. This change in predicted dynamics iscaused by a structural change in the drug binding site.
The loss of the hydrophobic interaction between theNNRTI and Y188 causes T229 to reorient, triggering asubtle rotation in b-12-13-14.20 This b-sheet is the plat-form upon which the thumb rests, so even a small rota-tion leads to a much larger displacement of the thumbaway from the connection subdomain (Fig. 4). Thismotion breaks several contacts in the model, whichreflects the breaking of several van der Waals contacts in
the structure. The ability of such a simple model of
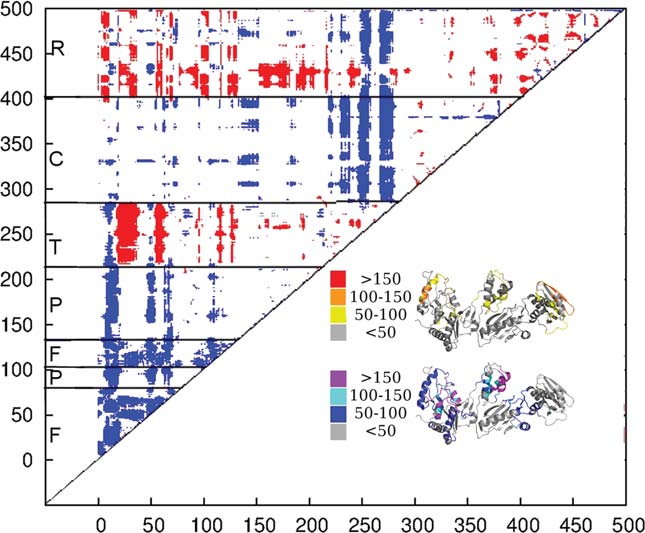
We calculated a matrix of significant differences between the inter-
motion to correctly predict the functional state from the
residue correlations of the inactive and preactive cluster according tothe Welch's t test. Residue pairs showing a significant difference between
structure suggests that the change in the predicted
the preactive and inactive cluster are colored. This difference can be
dynamics plays a vital role in the ability of the hydro-
more correlated (red) or more anticorrelated (blue) fashion. The resi-
phobic core mutants to offer resistances against first and
dues showing a strong difference in their inter-residue correlation tendto be regions of the protein which begin moving differently with
second generation NNRTIs. The hydrophobic core
regards to the rest of the structure. The number of residues changing
mutants were crystallized under inhibiting concentrations
their motion with respect to a single residue mapped onto the structure
of NNRTI, suggesting that the shift in dynamics alone in
of p66. The structure is colored by whether residues become more cor-
not enough to convey full drug resistance.16,20,47,48
related moving from the inactive to the preactive cluster (yellow,orange, and red) or more anticorrelated (blue, cyan, and purple).39
The answer can be found in the dynamics of the p51subunit. These results break cleanly into three groups
similar, with average pairwise RMSDs of 1.49 A˚. To
with the unliganded, active-cluster structures, and the
probe the differences in dynamics between the hydropho-
NNRTI bound structures and the 1RTJ unliganded struc-
bic core mutants in the preactive and inactive clusters,
ture forming their own clusters (Supporting Information
the unliganded structures were removed from the preac-
Fig. S3). The p51 subunit is required for RT activity, and
tive cluster and the clusters were compared again. It is
is necessary to propagate NNRTI-induced structural
clear that the NNRTI binding site moves differently in
rigidification.49 This suggests that p51 has a dynamic
the new subclusters, accompanied by significant differen-
role (in addition to its structural role) in activity and
ces in the internal motion of the thumb and connection
NNRTI binding disrupts this process. The structural
subdomain and the RNase H domain, restoring the
change from the inactive to the active p51 subunit is
hydrophobic core mutant structures to active-like inter-
very small (�1.2 A˚), suggesting that when an NNRTI
nal motions. Additionally, in the hydrophobic core
comes off, the conformational change to the active p51
mutant structures, the thumb subdomain moves in a
conformation can occur rapidly. This could also explain
more correlated fashion with respect to the RNase H
why a single non-NNRTI resistant mutant clustered with
domain, again like the active cluster (Supporting Infor-
the preactive cluster. This structure contains a trio of
mation Fig. S2).
nucleoside RT inhibitor mutants that have been shownto increase the resistance of certain combinations ofNNRTI-resistant mutations.50 This further suggests that
a reduction in drug binding efficiency is required along-side the change in dynamics.
To understand the function of RT, including how inhi-
Our work reveals two groups of drug-resistant muta-
bition and drug resistance works, it is first necessary to
tions by their effect on the dynamics of HIV-1 RT. These
understand both the native structure and the native
groups of mutations compare well with current theory of
dynamics. It has long been accepted that flexibility plays
drug resistance.12 The first group (V108I, Y181C, and
a crucial role in the proper function of RNA polymer-
Y188C) all feature mutations located deep within the
ases,42,43 and there have been many attempts to explain
hydrophobic core of the NNRTI-binding pocket, and are
the inhibition of HIV-1 RT by NNRTIs through changes
thought to cause drug resistance via a loss of an
Structure and Dynamics of HIV RT
interaction with aromatic rings.1,19,20,51,52 Our model
mode caused inhibition. It also suggests that changes in
predicts these mutations perturb the internal motions of
the overall topology of a protein have marked affects on
the thumb subdomain and the RNase H domain of HIV-
its activity and ability to bind ligands. Here, we consider
1 RT, restoring active state movement, without signifi-
both structure and dynamics must be considered: a sur-
cantly affecting the structure. Also in this group is one of
vey of many different structures of both wild type and
the most potent of the drug resistance mutations, K103N
drug resistant mutants suggests that it is both the change
bound to the first generation NNRTI Nevriapine. How-
in thumb domain motion and the relative thumb posi-
ever, all efavirenz-bound structures fall within the inac-
tion that causes inhibition, rather than the change in
tive cluster along with a wild-type structure bound to
motion alone. On the other hand, in ENM calculations
efavirenz. The fact that K103N falls within multiple clus-
there is no input other than the structure—geometry is
ters means that it cannot be conclusively stated whether
destiny—so cannot consider dynamics without structure.
it has an allosteric mechanism for resistance.
On the other hand, K101E and L100I have essentially
no effect on the structure of inhibited HIV-1 RT com-pared to wild type. These mutations, along with K103N,
The present work surveys the wealth of structural
are thought to make NNRTI binding unfavorable by
information available for RT, combining direct structural
either changing the shape of the binding pocket or
analysis with modeling of the complex's dynamics, using
blocking the inhibitor's entrance into the binding
a simple harmonic model. This reveals a wealth of previ-
pocket.14–16 The present work appears to validate this
ously hidden details about allosteric interactions due to
idea, showing that the protein assumes an inhibited
ligand and mutations. We propose a new model of
structure and dynamics upon binding of inhibitor.
NNRTI drug resistance whereby mutations to the hydro-
Recently, a mechanism for NNRTI inhibition was pro-
phobic core of the drug binding pocket cause dynamic
posed based on crystallographic data of HIV-1 RT bound
changes across the protein, restoring proper thumb and
to both the first general inhibitor Nevriapine and DNA.9
RNase H domain motions, and alter the motion of the
This model states that binding Nevriapine displaces the
polymerase domain to a more unliganded-like motion.
primer grip of HIV-1 RT and, when combined with the
This reveals ANM as a powerful bioinformatics tool for
hyperextended thumb conformation, moves the DNA
quickly probing the dynamics of known protein struc-
away from the polymerase active site. At the same time,
tures, allowing us to find novel allosteric interactions to
inhibitor binding distorts the dNTP binding site and
compliment and inform experiments.
shifts the relative position of the RNAse H and polymer-ase active sites. As a result, the post-translation complex
of the DNA-bound protein is bound to a catalyticallyinactive
We are grateful to the Center for Integrated Research
dynamics nearly indistinguishable from that of other
Computing at the University of Rochester for providing
inhibited structures, strongly suggesting a dynamic com-
the necessary computing systems and personnel to enable
ponent to the mispositioning of the DNA in the poly-
the research presented in this manuscript. We would also
merase active sight.
like to thank Dr. Tod Romo for help using LOOS and
The structural changes caused by the Y188C, Y181C,
performing the ENM calculations, and Dr. Ivet Bahar
and V108I mutations correspond to a previously discov-
and Dr. Carrie Dykes for advice in the preparation of the
ered allosteric network.4 This network appears via both
dynamic and structural analysis, suggesting that the allo-steric coupling of this network is encoded in the three-
dimensional structure of the protein, and furthermore
1. Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal
that perturbing other regions of this network causes
structure at 3.5 A˚ resolution of HIV-1 reverse transcriptase com-
global changes in the structure of the protein. This can
plexed with an inhibitor. Science 1992;256:1783-1790.
be seen in the case of the RIs, which bind in the network
2. Boyer PL, Ferris AL, Clark P, Whitmer J, Frank P, Tantillo C,
but are distant from the NNRTI binding site, and shift
Arnold E, Hughes SH. Mutational analysis of the fingers and palmsubdomains of human immunodeficiency virus type-1 (HIV-1)
the structure of nevirapine-bound K103N protein from
reverse transcriptase. J Mol Biol 1994;243:472-483.
the preactive cluster to the inactive cluster (Fig. 3;
3. Bahar I, Erman B, Jernigan RL, Atilgan AR, Covell DG. Collective
motions in HIV-1 reverse transcriptase: examination of flexibility
There have been many previous investigations using
and enzyme function. J Mol Biol 1999;285:1023-1037.
various types of molecular modeling to study HIV-1 RT.
4. Seckler JM, Barkley MD, Wintrode PL. Allosteric suppression of
Previous studies using elastic network modeling showed
HIV-1 reverse transcriptase structural dynamics upon inhibitorbinding. Biophys J 2011;100:144-153.
that NNRTI binding changed RT's global motions.3,45,53
5. Perno CF, Yarchoan R, Cooney DA, Hartman NR, Gartner S,
This work suggested that the change in the relative
Popovic M, Hao Z, Gerrard TL, Wilson YA, Johns DG, Broder S.
motion of the fingers and thumb subdomain in the first
Inhibition of human mmunodeficiency virus (HIV-1/HTLV-IIIBa-L)
J.M. Seckler et al.
replication in fresh and cultured human peripheral blood mono-
reverse transcriptase non-nucleoside inhibitor binding pocket: les-
cytes/macrophages by azidothymidine and related 20,30-dideoxynu-
sons for inhibitor design from a cluster analysis of many crystal
cleosides. J Exp Med 1988;168:1111-1125.
structures. J Med Chem 2009;52:6413-6420.
6. Shaw-Reid CA, Feuston B, Munshi V, Getty K, Krueger J, Hazuda
23. van Westen GJ, Wegner JK, Bender A, Ijzerman AP, van Vlijmen
DJ, Parniak MA, Miller MD, Lewis D. Dissecting the effects of DNA
HW. Mining protein dynamics from sets of crystal structures using
polymerase and ribonuclease H inhibitor combinations on HIV-1
"consensus structures". Protein Sci 2010;19:742-752.
reverse-transcriptase activities. Biochemistry 2005;44:1595-1606.
24. Grossfield A, Zuckerman DM. Quantifying uncertainty and sam-
7. Hang JQ, Li Y, Yang Y, Cammack N, Mirzadegan T, Klumpp K.
pling quality in biomolecular simulations. Annu Rep Comput
Substrate-dependent inhibition or stimulation of HIV RNase H
Chem 2009;5:23-48.
25. Romo TD, Grossfield A. Validating and improving elastic network
(NNRTIs). Biochem Biophys Res Commun 2007;352:341-350.
models with molecular dynamics simulations. Proteins 2011;79:
8. Radzio J, Sluis-Cremer N. Efavirenz accelerates HIV-1 reverse tran-
scriptase ribonuclease H cleavage, leading to diminished zidovudine
26. Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar
excision. Mol Pharmacol 2008;73:601-606.
I. Anisotropy of fluctuation dynamics of proteins with an elastic
9. Das K, Martinez SE, Bauman JD, Arnold E. HIV-1 reverse transcrip-
network model. Biophys J 2001;80:505-515.
tase complex with DNA and nevirapine reveals non-nucleoside inhi-
27. Temiz NA, Meirovitch E, Bahar I. Escherichia coli adenylate
bition mechanism. Nat Struct Mol Biol 2012;19:253-259.
kinase dynamics: comparison of elastic network model modes with
10. Ren J, Stammers DK. Structural basis for drug resistance mecha-
mode-coupling (15)N-NMR relaxation data. Proteins 2004;57:
nisms for non-nucleoside inhibitors of HIV reverse transcriptase.
Virus Res 2008;134:157-170.
28. Deriu MA, Soncini M, Orsi M, Patel M, Essex JW, Montevecchi
11. Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-
FM, Redaelli A. Anisotropic elastic network modeling of entire
Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK,
microtubules. Biophys J 2010;99:2190-2199.
Krogh-Jespersen K, Levy, RM, Hughes SH, Parniak MA, Arnold E.
29. Keskin O, Durell SR, Bahar I, Jernigan RL, Covell DG. Relating
HIV-1 reverse transcriptase structure with RNase H inhibitor dihy-
molecular flexibility to function: a case study of tubulin. Biophys J
droxy benzoyl naphthyl hydrazone bound at a novel site. ACS
Chem Biol 2006;1:702-712.
30. Mao Y. Dynamical basis for drug resistance of HIV-1 protease.
12. Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA,
BMC Struct Biol 2011;11:31.
Hughes SH, Arnold E. Structure and function of HIV-1 reverse
31. Wang Y, Rader AJ, Bahar I, Jernigan RL. Global ribosome motions
transcriptase: molecular mechanisms of polymerization and inhibi-
revealed with elastic network model. J Struct Biol 2004;147:302-314.
tion. J Mol Biol 2009;385:693-713.
32. Leioatts N, Romo TD, Grossfield A. Elastic network models are
13. Miller CD, Crain J, Tran B, Patel N. Rilpivirine: a new addition to
robust to variations in formalism. J Chem Theory Comput 2012;8:
the anti-HIV-1 armamentarium. Drugs Today (Barc) 2011;47:5-15.
14. Hsiou Y, Ding J, Das K, Clark AD, Jr, Boyer PL, Lewi P, Janssen PA,
33. Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr, Brice MD,
Kleim JP, Rosner M, Hughes SH, Arnold E. The Lys103Asn muta-
Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein
tion of HIV-1 RT: a novel mechanism of drug resistance. J Mol Biol
Data Bank: a computer-based archival file for macromolecular
structures. J Mol Biol 1977;112:535-542.
15. Ren J, Nichols CE, Chamberlain PP, Weaver KL, Short SA, Chan
34. Woodcock HL, Zheng W, Ghysels A, Shao Y, Kong J, Brooks BR.
JH, Kleim JP, Stammers DK. Relationship of potency and resilience
Vibrational subsystem analysis: a method for probing free energies
to drug resistant mutations for GW420867X revealed by crystal
and correlations in the harmonic limit. J Chem Phys 2008;129:
structures of inhibitor complexes for wild-type, Leu100Ile, Lys101-
Glu, and Tyr188Cys mutant HIV-1 reverse transcriptases. J Med
35. Hess B. Convergence of sampling in protein simulations. Phys Rev
E Stat Nonlin Soft Matter Phys 2002;65:031910.
16. Ren J, Nichols CE, Chamberlain PP, Weaver KL, Short SA,
36. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and
Stammers DK. Crystal structures of HIV-1 reverse transcriptases
display of genome-wide expression patterns. Proc Natl Acad Sci
mutated at codons 100, 106 and 108 and mechanisms of resistance
to non-nucleoside inhibitors. J Mol Biol 2004;336:569-578.
37. Van Wynsberghe AW, Cui Q. Interpreting correlated motions using
17. Sarafianos SG, Das K, Hughes SH, Arnold E. Taking aim at a mov-
normal mode analysis. Structure 2006;14:1647-1653.
ing target: designing drugs to inhibit drug-resistant HIV-1 reverse
38. Fisher RA. Frequency distribution of the values of the correlation
transcriptases. Curr Opin Struct Biol 2004;14:716-730.
coefficient in samples of an indefinitely large population. Biome-
18. Hsiou Y, Das K, Ding J, Clark AD, Jr, Kleim JP, Rosner M, Winkler
I, Riess G, Hughes SH, Arnold, E. Structures of Tyr188Leu mutant
39. Welch BL. The generalisation of student's problems when several
and wild-type HIV-1 reverse transcriptase complexed with the non-
different population variances are involved. Biometrika 1947;34:
nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design
feature for reducing drug resistance. J Mol Biol 1998;284:313-323.
40. Romo TD, Grossfield A. LOOS: an extensible platform for the struc-
19. Ren J, Esnouf R, Hopkins A, Ross C, Jones Y, Stammers D, Stuart
tural analysis of simulations. Conf Proc IEEE Eng Med Biol Soc
D. The structure of HIV-1 reverse transcriptase complexed with 9-
chloro-TIBO: lessons for inhibitor design. Structure 1995;3:915-926.
41. Kurle SN, Gangakhedkar RR, Sen S, Hayatnagarkar SS, Tripathy SP,
20. Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart
Paranjape RS. Emergence of NNRTI drug resistance mutations after
DI, Stammers DK. Structural mechanisms of drug resistance for
single-dose nevirapine exposure in HIV type 1 subtype C-infected
mutations at codons 181 and 188 in HIV-1 reverse transcriptase
infants in India. AIDS Res Hum Retroviruses 2007;23:682-685.
and the improved resilience of second generation non-nucleoside
42. Steitz TA. Visualizing polynucleotide polymerase machines at work.
inhibitors. J Mol Biol 2001;312:795-805.
EMBO J 2006;25:3458-3468.
21. O'Brien SE, Brown DG, Mills JE, Phillips C, Morris G. Computa-
43. Svetlov V, Nudler E. Macromolecular micromovements: how RNA
tional tools for the analysis and visualization of multiple protein–
polymerase translocates. Curr Opin Struct Biol 2009;19:701-707.
ligand complexes. J Mol Graph Model 2005;24:186-194.
44. Shen L, Shen J, Luo X, Cheng F, Xu Y, Chen K, Arnold E, Ding J,
22. Paris KA, Haq O, Felts AK, Das K, Arnold E, Levy RM. Conforma-
Jiang H. Steered molecular dynamics simulation on the binding of
tional landscape of the human immunodeficiency virus type 1
NNRTI to HIV-1 RT. Biophys J 2003;84:3547-3563.
Structure and Dynamics of HIV RT
45. Temiz NA, Bahar I. Inhibitor binding alters the directions of
and effective against wild-type and drug-resistant HIV-1 variants.
domain motions in HIV-1 reverse transcriptase. Proteins 2002;49:
J Med Chem 2004;47:2550-2560.
52. Das K, Ding J, Hsiou Y, Clark AD, Jr, Moereels H, Koymans L,
46. Zhou Z, Madrid M, Evanseck JD, Madura JD. Effect of a bound
Andries K, Pauwels R, Janssen PA, Boyer PL, Clark P, Smith RH, Jr,
non-nucleoside RT inhibitor on the dynamics of wild-type and
Kroeger Smith MB, Michejda CJ, Hughes SH, Arnold E. Crystal
mutant HIV-1 reverse transcriptase. J Am Chem Soc 2005;127:
structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1
RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-
47. Chamberlain PP, Ren J, Nichols CE, Douglas L, Lennerstrand J,
resistant mutant. J Mol Biol 1996;264:1085-1100.
Larder BA, Stuart DI, Stammers DK. Crystal structures of Zidovu-
53. Bakan A, Bahar I. The intrinsic dynamics of enzymes plays a domi-
dine- or Lamivudine-resistant human immunodeficiency virus type
nant role in determining the structural changes induced upon
1 reverse transcriptases containing mutations at codons 41, 184, and
inhibitor binding. Proc Natl Acad Sci USA 2009;106:14349-14354.
215. J Virol 2002;76:10015-10019.
54. Hsiou Y, Ding J, Das K, Clark AD, Jr, Hughes SH, Arnold E. Struc-
48. Ren J, Milton J, Weaver KL, Short SA, Stuart DI, Stammers DK.
ture of unliganded HIV-1 reverse transcriptase at 2.7 A resolution:
Structural basis for the resilience of efavirenz (DMP-266) to drug
implications of conformational changes for polymerization and
resistance mutations in HIV-1 reverse transcriptase. Structure 2000;
inhibition mechanisms. Structure 1996;4:853-860.
55. Sarafianos SG, Clark AD, Jr, Das K, Tuske S, Birktoft JJ, Ilankumaran
49. Braz VA, Holladay LA, Barkley MD. Efavirenz binding to HIV-1
P, Ramesha AR, Sayer JM, Jerina DM, Boyer PL, Hughes SH, Arnold
reverse transcriptase monomers and dimers. Biochemistry 2010;49:
E. Structures of HIV-1 reverse transcriptase with pre- and post-
translocation AZTMP-terminated DNA. EMBO J 2002;21:6614-6624.
50. Wang J, Liang H, Bacheler L, Wu H, Deriziotis K, Demeter LM,
56. Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mecha-
Dykes C. The non-nucleoside reverse transcriptase inhibitor efavir-
nism of inhibition of HIV-1 reverse transcriptase by non-nucleoside
enz stimulates replication of human immunodeficiency virus type 1
inhibitors. Nat Struct Biol 1995;2:303-308.
harboring certain non-nucleoside resistance mutations. Virology
57. Ren J, Esnouf R, Garman E, Somers D, Ross C, Kirby I, Keeling J,
Darby G, Jones Y, Stuart D, Stammers D. High resolution structures
51. Das K, Clark AD, Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans
of HIV-1 RT from four RT-inhibitor complexes. Nat Struct Biol
LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B,
Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R,
58. Lindberg J, Sigurdsson S, Lowgren S, Andersson HO, Sahlberg C,
De Bethune MP, Boyer PL Clark P, Hughes SH, Janssen PA, Arnold
Noreen R, Fridborg K, Zhang H, Unge T. Structural basis for the
E. Roles of conformational and positional adaptability in structure-
inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721
based design of TMC125-R165335 (etravirine) and related non-
towards the HIV-1 RT K103N mutant. Eur J Biochem 2002;269:
nucleoside reverse transcriptase inhibitors that are highly potent
Source: http://membrane.urmc.rochester.edu/sites/default/files/papers/proteins_2013.pdf
Florida State College at Jacksonville Assessment Data by Section 2011-2012 Academic Success Center Student Learning Outcomes/Objectives SLO 2: Effective Advising Services The Academic Success Centers effectively provide advising services for developmental students seeking academic and personal assistance at each campus and center.
This compilation is for general information only and not to be used for profit or a substitute for yourtraining. Consult a professional. Please respect IP & copyright Laws. Slight variations may occur for layout purposes.Errors and omissions are not intentional, Your Host & Hostess, Chief Dave "Passing Wind" & Faith "D'Sagwagon" Substances: World Anti-Doping Code:


