
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Rusynlab.com
Carcinogenesis vol.21 no.12 pp.2141–2145, 2000
Expression of base excision repair enzymes in rat and mouse liver
is induced by peroxisome proliferators and is dependent upon
carcinogenic potency
Ivan Rusyn1,2, Mikhail F.Denissenko4, Victoria A.Wong5,
The major mechanisms that have been proposed for peroxi-
Byron E.Butterworth5, Michael L.Cunningham6,
Patricia B.Upton1, Ronald G.Thurman2,3 and
increased cell proliferation leading to promotion of spontan-
eously initiated lesions; (ii) oxidative stress, resulting from
disproportionate increases in the levels of oxidants. However,
Department of Environmental Sciences and Engineering, 2Curriculum in
recent observations that oxidant-dependent activation of the
Toxicology, and 3Department of Pharmacology, University of NorthCarolina at Chapel Hill, Chapel Hill, NC 27599, 4BD PharMingen, San
transcription factor NF-κB plays a critical role in increased
Diego, CA 92121, 5Chemical Industry Institute of Toxicology, Research
hepatocyte proliferation caused by WY-14,643 (5) and cipro-
Triangle Park, NC 27709 and 6National Institute of Environmental Health
fibrate (6) suggest that these mechanisms may not be mutually
Sciences, National Institutes of Health, Research Triangle Park, NC 27709,
exclusive and oxidants may be involved in signaling increased
cell proliferation.
7To whom correspondence should be addressed at: Department of
It has been suggested that reactive oxygen species play a
Environmental Sciences and Engineering, CB 7400, University of NorthCarolina at Chapel Hill, Chapel Hill, NC 27599-7400, USA
role in the initiation and promotion steps of carcinogenesis
induced by peroxisome proliferators. Recently, direct evidenceof rapid peroxisome proliferator-induced generation of
Elevated and sustained cell replication, together with a
hydroxyl radicals
in vivo has been presented (7). Indeed,
decrease in apoptosis, is considered to be the main mechan-
ism of hepatic tumor promotion due to peroxisome pro-
production of reactive oxygen species may lead to DNA
liferators. In contrast, the role of oxidative stress and
damage via hydroxyl radicals and products of lipid peroxida-
DNA damage in the carcinogenic mechanism is less well
tion. Oxidative stress is hypothesized to be a common pathway
understood. In view of possible induction of DNA damage
for many non-genotoxic chemical carcinogens (8). However,
by peroxisome proliferators, DNA repair mechanisms may
the role of oxidative stress has been questioned. Indeed, when
be an important factor to consider in the mechanism of
compared with direct DNA damaging agents, the magnitude
action of these compounds. Here, the ability of peroxisome
of response following carcinogenic exposures to chemicals
proliferators to induce expression of base excision repair
thought to work through oxidants has been small. Several
enzymes was examined. WY-14,643, a potent carcinogen,
attempts to assess oxidative DNA damage by peroxisome
increased expression of several base excision DNA repair
proliferators using direct measurement of adducts produced
enzymes in a dose- and time-dependent manner. Import-
equivocal results (9,10). Moreover, the artifactual formation
antly, expression of enzymes that do not repair oxidative
of oxidized base damage due to artifactual auto-oxidation
DNA damage was not changed. Moreover, less potent
reactions in assays requiring extraction of DNA has plagued
members of the peroxisome proliferator group had much
this experimental approach (11).
weaker or no effects on expression of DNA repair enzymes
On the other hand, it is known that DNA repair enzymes
when compared with WY-14,643. Collectively, these data
are induced both
in vivo and
in vitro by oxidative stress (12).
suggest that DNA base excision repair may be an important
Several DNA repair genes involved in oxidative damage have
factor in peroxisome proliferator-induced carcinogenesis
been identified and it is believed that the predominant pathway
and that induction of DNA repair might provide further
used for removal of oxidized bases is the base excision repair
evidence supporting a role of oxidative DNA damage by
(BER) pathway. Several proteins are involved in this multi-
step repair process (12). For instance, the primary pathwayfor removal of 8-hydroxydeoxyguanosine (8-OH-dG) appearsto be OGG1, a glycosylase/lyase, which excises this adduct
from DNA and cleaves 3⬘ to AP (abasic) sites, leaving a 3⬘-cleaved AP site (13). The DNA is then cleaved 5⬘ to the AP
Peroxisome proliferators are a diverse group of chemicals
site by AP endonuclease (APE), the gap is filled by polymerase
and therapeutic agents. In rodents these compounds cause
β (Pol β) and the newly synthesized DNA is sealed by ligase
hepatomegaly, proliferation of peroxisomes in hepatic paren-
(14). While BER is considered the main pathway for oxidative
chymal cells and marked increases in the activity of enzymesrequired for peroxisomal
DNA damage, nucleotide excision repair and long patch repair
β-oxidation of fatty acids (1). These
changes persist at steady-state levels as long as peroxisome
have also been shown to remove oxidative damage from DNA
proliferators are administered. Long-term exposure results in
(15). Importantly, expression of enzymes that participate in
the development of liver tumors in rodents (2). Rodents are
these processes may be induced following increased production
much more sensitive to the effects of peroxisome proliferators
of reactive oxygen species (16) or chemical exposure (17).
than dogs, non-human primates or humans. This difference in
No previous reports, however, have described changes in
sensitivity appears to be due to differences in the PPARα
DNA repair enzymes associated with peroxisome proliferator-
receptor and its response elements (reviewed in ref. 3). Most
induced carcinogenesis. Since BER is a major mechanism for
reviews of peroxisome proliferators conclude that humans are
removal of oxidative lesions from DNA (18), we investigated
at little or no risk for cancer from exposure to these agents (4).
the expression of several DNA glycosylases, APE, DNA
Oxford University Press
I.Rusyn et al.
polymerases and ligases in livers of rats (Fisher 344) and mice(C57Bl/6) treated with peroxisome proliferators. To assemblea comprehensive set of liver tissues, a combination of samplesfrom several different studies performed at the University ofNorth Carolina at Chapel Hill, Chemical Industry Instituteof Toxicology, or by the National Toxicology Program hasbeen used.
WY-14,643 causes a dose-dependent increase in expression
of DNA BER enzymes in rodent liver
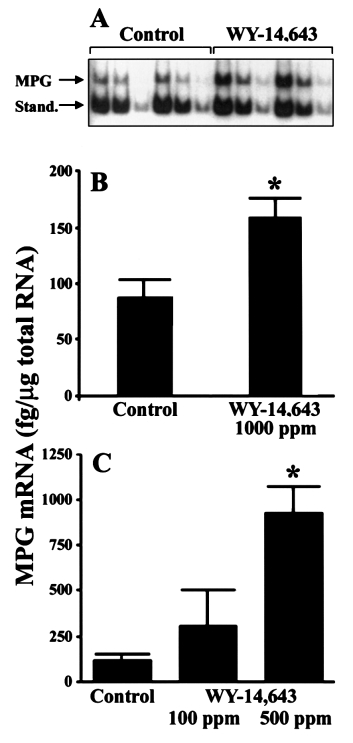
Mammalian N-methylpurine-DNA glycosylase (MPG) hasbroad substrate specificity and primarily is capable of hydro-lysis of 3-methyladenine. It has also been shown to hydrolyze1,N6-ethenoadenine in vitro and weak activity towards 8-OH-dG has been reported (19). A quantitative reverse transcriptasePCR assay (19) was used to test the hypothesis that peroxisomeproliferators induce expression of MPG in rat liver. The useof in vitro synthesized reference RNA allowed quantitation ofthe PCR products (MPG and standard) after gel electrophoresisand autoradiography (Figure 1A). The effect of a 21 daytreatment with WY-14,643 (1000 p.p.m.) on MPG mRNA inwhole rat liver is shown in Figure 1B. In livers of animals feda regular chow diet, expression of MPG mRNA was low (89
⫾ 16 fg/µg total RNA), however, it was elevated 2-fold aftertreatment with WY-14,643 (164 ⫾ 21 fg/µg total RNA).
Moreover, a dose-dependent increase in expression of MPGwas observed in rats treated with WY-14,643 for 90 days(Figure 1C). The amount of MPG mRNA was 2- or 6.5-foldhigher in livers of rats treated with 100 or 500 p.p.m. WY-14,643, respectively, than in control animals. Collectively,these results support the hypothesis that potent peroxisomeproliferators (i.e. WY-14,643) may inflict DNA damage andthat levels of DNA repair gene expression could be used toestablish a gene response profile after exposure to thesechemicals.
Next, we used a recently developed multi-probe RNase
protection assay for BER enzymes. This approach distinguishesthe presence of multiple expressed DNA repair genes simultan-eously from a single sample, which allows comparative analysisof different mRNA products both within and between samples.
This is a highly sensitive and specific approach for detection
Fig. 1. MPG mRNA is induced in rat liver by dietary feeding of WY-
and quantitation of gene expression at the mRNA level. It
14,643. (A) An autoradiogram of PCR products of native mRNA and
synthetic MPG RNA (a kind gift of Dr S. Mitra, University of Texas,
should be noted, however, that the RNase protection assay
Galveston, TX) after EcoRI digestion is shown. Fisher 344 rats were fed
template sets (a kind gift of BD PharMingen, San Diego, CA)
either control diet (NIH-07) or diet containing WY-14,643 (1000 p.p.m. for
used here for rat and mouse tissues were under development
21 days). Total cellular RNA was isolated using a QuickPrep extraction kit
and differed slightly in the composition of DNA repair genes
(Amersham Pharmacia Biotech) and RT–PCR was performed as describedin Roy et al. (19). Three 1:2 serial dilutions of each sample mixture
evaluated. Total mRNA was isolated from livers of rats fed
containing 2 µg total cellular RNA and 540 fg synthetic RNA standard were
control or WY-14,643 (1000 p.p.m.)-containing diet for up to
made, amplified by PCR and resolved on 6% polyacrylamide gels. The 471
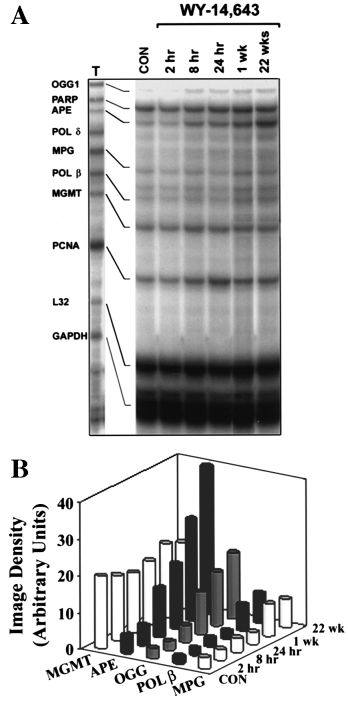
22 weeks. A time-dependent 3- to 12-fold increase in mRNA
bp product of cellular MPG amplification and 366 bp product of reference
for OGG1, APE, MPG and Pol β was observed (Figure 2).
RNA amplification after enzymatic cleavage of the 105 bp non-radioactive
Importantly, expression of several enzymes that are not related
fragment are indicated. Representative autoradiogram. (B) Data shown are
results of densitometry analysis of images from the experiment detailed in
to oxidative DNA damage (e.g. O6-methylguanine-DNA
(A). (C) As above but rats were given 0 (Control), 100 or 500 p.p.m. WY-
methyltransferase and polymerase δ) was not changed. It
14,643 for 90 days. Data reported as means ⫾ SEM from four separate
should be noted that our findings of an increased expression
experiments. Asterisks (*) denote statistical differences (P ⬍ 0.05) from the
of mRNA for BER proteins were corroborated recently when
control group by one way ANOVA and Student–Newman–Keuls post hoctest.
it was reported that Pol β and APE protein levels wereincreased markedly after treatment with WY-14,643 for 6days (20).
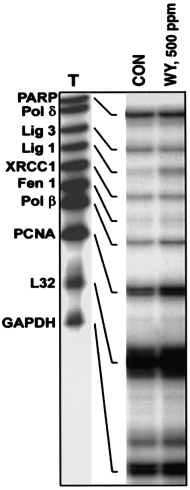
patch' repair pathway, was induced in mouse liver after dietary
The single nucleotide BER pathway is a favorable mechan-
treatment with WY-14,643 (500 p.p.m.) for 7 days (Figure 3).
ism for removal of oxidized bases and is dependent upon the
Similar to what was observed in rats, WY-14,643 (500 p.p.m.)
interaction between DNA Pol β and DNA ligase I (21). Indeed,
caused an 3-fold increase in mRNA for OGG1, TDG, APE,
only ligase I, but not ligase III or other enzymes of the ‘long
MPG and Nth1 in mouse liver (data not shown). These findings
Peroxisome proliferators induce DNA repair
Fig. 3. Expression of ligase I is induced in mouse liver by WY-14,643.
Mice (C57Bl/6) were fed a diet containing the potent carcinogen WY-
14,643 (WY, 500 p.p.m.) for 7 days. Total RNA was isolated from liver
samples and used in the RNase protection assay as detailed in Figure 2.
Representative autoradiograms from three separate experiments.
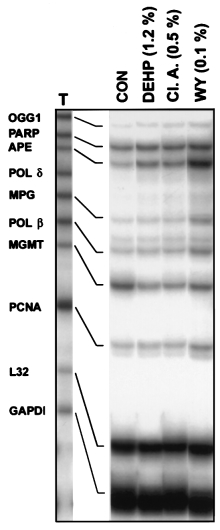
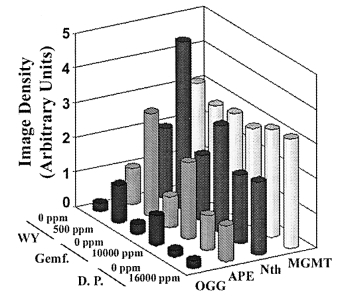
carcinogens from this group of compounds have similar effectson DNA repair enzymes, rats were fed WY-14,643 or theweaker carcinogens di(2-ethyhexyl)phthalate (DEHP) and clo-fibric acid for 22 weeks. Both DEHP (12000 p.p.m.) andclofibric acid (5000 p.p.m.) increased expression of mRNAfor OGG1, APE, MPG and Pol β by 2- to 3-fold, however,
Fig. 2. WY-14,643 causes a time-dependent increase in expression of base
these effects fell short of the profound changes caused by
excision DNA repair enzymes in rat liver. (A) Total RNA was isolated from
WY-14,643 (1000 p.p.m.) (Figure 4). It should be noted that
livers of rats treated with WY-14,643 (1000 p.p.m.) for up to 22 weeks
since WY-14,643, DEHP and clofibric acid cause a similar
using a QuickPrep extraction kit (Amersham Pharmacia Biotech) anddissolved in RNase-free water. Expression of base excision DNA repair
initial increase in cell proliferation, the differences in expres-
enzymes was analyzed with an RNase protection assay using rodent multi-
sion of DNA repair enzymes observed here are not due to a
nucleotide RNA probe template sets (a generous gift of BD PharMingen).
rapid growth of liver mass. Furthermore, when WY-14,643
Riboprobes were synthesized in the presence of [32P]dUTP to yield labeled
(1000 p.p.m.) and DEHP (12000 p.p.m.) were administered to
antisense RNA probes. The RNase protection assay was performed on 40
rats for 7 days, induction of OGG1 APE and Nth1 was
µg of individual RNA samples using a RiboQuant multi-probe RNaseprotection assay kit (BD PharMingen). Protected fragments were separated
observed only in WY-14,643-treated animals (data not shown).
on QuickPoint nucleic acid separation gels (Novex, San Diego, CA), dried
Similar effects were observed with gemfibrozil, another potent
and exposed to X-ray film. Representative autoradiogram from three
rodent carcinogen, but not with the weak carcinogen dibutyl
separate experiments. (B) Data shown are results of densitometry analysis of
phthalate (Figure 5). Specifically, both WY-14,643 (500 p.p.m.)
images from the experiment detailed in (A). The intensity of protectedbands was quantified using an image analyzer and normalized to the
and gemfibrozil (16000 p.p.m.) administered to rats for 90
intensity of housekeeping genes L32 and GAPDH.
days increased expression of DNA BER enzymes, while dibutylphthalate (10000 p.p.m.) had no effect.
are important because they show that WY-14,643 induces
It has been hypothesized that the hepatocarcinogenicity of
expression of the whole pathway responsible for the repair of
peroxisome proliferators is due to oxidative stress. At least
oxidative DNA damage, DNA glycosylases, APE, Pol β and
two possible sources of oxidants following administration of
peroxisome proliferators have been proposed: (i) peroxisomalacyl CoA oxidase in parenchymal cells; (ii) NADPH oxidase
Potent carcinogens are more potent inducers of BER
in Kupffer cells (reviewed in ref. 22). The data presented here
enzymes gene expression
are important for mechanistically based risk assessment of
WY-14,643 is one of the most potent carcinogens among
peroxisome proliferators for several reasons. First, further
the peroxisome proliferators. To test whether less potent
evidence supporting the role of oxidative DNA damage in the
I.Rusyn et al.
mechanism of action of peroxisome proliferators is provided.
Specifically, we suggest that DNA BER may be an importantfactor in peroxisome proliferator-induced carcinogenesis andthat induction of DNA repair reflects an increase in oxidizedbases following treatment with these compounds. The smalland inconsistent increases in oxidative DNA damage observedin previous studies may be due to an inability to observe realincreases due to the artifactual formation of oxidative DNAdamage during DNA isolation. Most of the studies havereported 8-OH-dG values in control rat livers of 2 per 10–5bp. These values can be dramatically reduced through the useof free radical trapping reagents and antioxidants (23, 24).
Reductions in artifactual 8-OH-dG are likely to increase thelevel of detectable oxidative DNA damage resulting fromexposure to peroxisome proliferators.
Second, overexpression of DNA repair enzymes per se may
play an important role in the mechanism of action of peroxi-some proliferators. For instance, Pol β is normally expressedat low levels and has high infidelity in replicating DNA (25).
Recently it was shown that overexpression of Pol β may resultin increased spontaneous mutagenesis (26). Moreover, it washypothesized that an excess of Pol β may disrupt functions ofother DNA polymerases by introducing illegitimate deoxyri-bonucleotides or mutagenic base analogs like those producedby oxidative stress (27).
In addition, APE (Ref-1) is a multifunctional protein that
stimulates DNA binding by a number of transcription factors,such as NF-κB and AP-1 (28). Interestingly, peroxisomeproliferators increase the activity of NF-κB in rodent liver andit was hypothesized that NF-κB activation plays a role in
Fig. 4. Chronic treatment with peroxisome proliferators that are potent
increased cell proliferation induced by these compounds
carcinogens induces increases in DNA BER enzymes expression. TotalRNA was isolated from livers of rats treated with DEHP (1.2% w/w),
(reviewed in ref. 22). Therefore, the marked increase in
clofibric acid (Cl. A., 0.5% w/w) or WY-14,643 (WY, 0.1% w/w) for 22
APE expression observed here might be important for the
weeks and used in the RNase protection assay as detailed in Figure 2.
promotional activity of peroxisome proliferators. On the other
Representative autoradiogram from three separate experiments.
hand, APE is known to regulate transactivation of p53 (29).
This transactivation only requires the C-terminus and has beensuggested to delay the G1/S transition and enhance BER (29).
Given that p53 has been shown to enhance BER (30), it ispossible that up-regulation of APE expression by peroxisomeproliferators leads to enhanced p53-dependent BER. Thesignificance of APE transactivation of the pro-apoptotic func-tions of p53 is unclear, since peroxisome proliferators areknown to decrease apoptosis in liver (31).
An additional concern is that DNA repair enzymes could
be up-regulated unevenly, so that a state of imbalanced DNArepair may exist. Indeed, imbalanced DNA repair may lead toformation of both mutagenic and clastogenic lesions. In thecase of oxidative DNA damage, if glycosylase and APE areoverabundant (relative to DNA polymerase or DNA ligase) thenDNA strand breaks might accumulate that could consequentlyinfluence cell viability and induce chromosomal damage (32).
Whether peroxisome proliferators cause imbalanced repair hasyet to be determined.
Finally, since current risk assessment of peroxisome pro-
liferators is based on important differences between humansand rodents in expression of PPARα (reviewed in ref. 3), the
Fig. 5. Sub-chronic treatment with the potent carcinogens WY-14,643 and
approach of analyzing expression of DNA repair enzymes can
gemfibrozil, but not with the weak carcinogen dibutyl phthalate, increasesexpression of DNA excision repair enzymes. Total RNA was isolated from
be used in further studies to assess whether or not BER
livers of rats treated with WY-14,643 (WY, 500 p.p.m.), dibutyl phthalate
enzymes, as a biomarker of oxidative stress, are induced by
(D.P., 10000 p.p.m.) or gemfibrozil (Gemf., 16000 p.p.m.) for 90 days and
WY-14,643 in PPARα knockout mice.
used in the RNase protection assay as detailed in Figure 2. Data shown are
In summary, the results of this study provide new information
results of densitometry analysis and the intensity of protected bands was
that supports a role of oxidative stress as a mechanism of
quantified using an image analyzer and normalized to the intensity ofhousekeeping genes L32 and GAPDH.
carcinogenesis for peroxisome proliferators. It demonstrates a
Peroxisome proliferators induce DNA repair
8-hydroxyguanine in DNA, its repair and OGG1 mRNA in rat lungs after
clear induction of DNA repair pathways associated with
intratracheal administration of diesel exhaust particles. Carcinogenesis,
oxidative DNA damage that is related to the dose and length
of exposure, as well as the potency of the peroxisome pro-
17. Holt,S., Roy,G., Mitra,S., Upton,P.B., Bogdanffy,M.S. and Swenberg,J.A.
liferator for inducing hepatic carcinogenesis.
(2000) Deficiency of N-methylpurine-DNA-glycosylase expression in
nonparenchymal cells, the target cell for vinyl chloride and vinyl fluoride.
Mutat. Res., 460, 105–115.
18. Demple,B. and Harrison,L. (1994) Repair of oxidative damage to DNA:
enzymology and biology. Annu. Rev. Biochem., 63, 915–948.
This work was supported, in part, by grants from the NIEHS (ES-09785 and
19. Roy,G., Roy,R. and Mitra,S. (1997) Quantitative reverse transcriptase
polymerase chain reaction for measuring the N-methylpurine-DNA
glycosylase mRNA level in rodent cells. Anal. Biochem., 246, 45–51.
20. Holmes,E.W., Bingham,C.M., Keshavarzian,A. and Cunningham,M.L.
(2000) Hepatic expression of DNA polymerase β, Ref-1, Bcl-2 and Bax
1. Lake,B.G. and Reddy,J.K. (1995) Working Group on Peroxisome
proteins in peroxisome proliferator-treated rats and hamsters. Toxicol. Sci.,
Proliferation: Peroxisome Proliferation and its Role in Carcinogenesis.
International Agency for Research on Cancer. (IARC), Lyon.
21. Dimitriadis,E.K.,
2. Reddy,J.K. and Lalwani,N.D. (1983) Carcinogenesis by hepatic peroxisome
Lewis,M.S. and Wilson,S.H. (1998) Thermodynamics of human DNA
proliferators: evaluation of the risk of hypolipidemic drugs and industrial
ligase I trimerization and association with DNA polymerase beta. J. Biol.
plasticizers to humans. CRC Crit. Rev. Toxicol., 12, 1–58.
Chem., 273, 20540–20550.
3. Gonzalez,F.J., Peters,J.M. and Cattley,R.C. (1998) Mechanism of action
22. Rusyn,I., Rose,M.L., Bojes,H.K. and Thurman,R.G. (2000) Novel role of
of the nongenotoxic peroxisome proliferators: role of the peroxisome
oxidants in the molecular mechanism of action of peroxisome proliferators.
proliferator-activated receptor alpha. J. Natl Cancer Inst., 90, 1702–1709.
Antioxidants Redox Signal., 2, 607–621.
4. Newman,T.B. and Hulley,S.B. (1996) Carcinogenicity of lipid-lowering
23. Helbock,H.J., Beckman,K.B., Shigenaga,M.K., Walter,P.B., Woodall,A.A.,
drugs. J. Am. Med. Assoc., 275, 55–60.
Yeo,H.C. and Ames,B.N. (1998) DNA oxidation matters: the HPLC-
5. Rusyn,I., Tsukamoto,H. and Thurman,R.G. (1998) WY-14,643 rapidly
electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-
activates nuclear factor κB in Kupffer cells before hepatocytes.
guanine. Proc. Natl Acad. Sci. USA, 95, 288–293.
24. Nakamura,J., La,D.K. and Swenberg,J.A. (2000) 5⬘-Nicked apurinic/
6. Nilakantan,V., Spear,B.T. and Glauert,H.P. (1998) Liver-specific catalase
apyrimidinic sites are resistant to beta-elimination by beta-polymerase and
expression in transgenic mice inhibits NF-κB activation and DNA synthesis
are persistent in human cultured cells after oxidative stress. J. Biol. Chem.,
induced by the peroxisome proliferator ciprofibrate. Carcinogenesis, 19,
25. Kunkel,T.A. (1985) The mutational specificity of DNA polymerase-beta
during in vitro DNA synthesis. Production of frameshift, base substitution,
Thurman,R.G. (1999) Phthalates rapidly increase reactive oxygen species
and deletion mutations. J. Biol. Chem., 260, 5787–5796.
in vivo. Free Radic. Biol. Med., 27, S148.
26. Canitrot,Y., Cazaux,C., Frechet,M., Bouayadi,K., Lesca,C., Salles,B. and
8. Klaunig,J.E., Xu,Y., Isenberg,J.S., Bachowski,S., Kolaja,K.L., Jiang,J.,
Hoffmann,J.S. (1998) Overexpression of DNA polymerase beta in cell
Stevenson,D.E. and Walborg,E.F. (1998) The role of oxidative stress in
results in a mutator phenotype and a decreased sensitivity to anticancer
chemical carcinogenesis. Environ. Health Perspect., 106, 289–295.
drugs. Proc. Natl Acad. Sci. USA, 95, 12586–12590.
9. Kasai,H., Okada,Y., Nishimura,S., Rao,M.S. and Reddy,J.K. (1989)
27. Canitrot,Y., Frechet,M., Servant,L., Cazaux,C. and Hoffmann,J.S. (1999)
Formation of 8-hydroxydeoxyguanosine in liver DNA of rats following
Overexpression of DNA polymerase beta: a genomic instability enhancer
long-term exposure to a peroxisome proliferator. Cancer Res., 49, 2603–
process. FASEB J., 13, 1107–1111.
28. Xanthoudakis,S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) Redox
10. Hegi,M.E., Ulrich,D., Sagelsdorff,P., Richter,C. and Lutz,W.K. (1990) No
activation of Fos-Jun DNA binding activity is mediated by a DNA repair
measurable increase in thymidine glycol or 8-hydroxydeoxyguanosine in
enzyme. EMBO J., 11, 3323–3335.
liver DNA of rats treated with nafenopin or choline-devoid low-methionine
29. Gaiddon,C., Moorthy,N.C. and Prives,C. (1999) Ref-1 regulates the
diet. Mutat. Res., 238, 325–329.
transactivation and pro-apoptotic functions of p53 in vivo. EMBO J., 18,
11. Cadet,J., D'Ham,C., Douki,T., Pouget,J.P., Ravanat,J.L. and Sauvaigo,S.
(1998) Facts and artifacts in the measurement of oxidative base damage
30. Offer,H., Wolkowicz,R., Matas,D., Blumenstein,S., Livneh,Z. and Rotter,V.
to DNA. Free Radic. Res, 29, 541–550.
(1999) Direct involvement of p53 in the base excision repair pathway of
12. Bohr,V.A. and Dianov,G.L. (1999) Oxidative DNA damage processing in
the DNA repair machinery. FEBS Lett., 450, 197–204.
nuclear and mitochondrial DNA. Biochimie, 81, 155–160.
31. Marsman,D.S., Goldsworthy,T.L. and Popp,J.A. (1992) Contrasting
13. Rosenquist,T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and
hepatocytic peroxisome proliferation, lipofuscin accumulation and cell
characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc.
replication for the hepatocarcinogens WY-14,643 and clofibric acid.
Natl Acad. Sci. USA, 94, 7429–7434.
14. Tomkinson,A.E. and Mackey,Z.B. (1998) Structure and function of
32. Posnick,L.M. and Samson,L.D. (1999) Imbalanced base excision repair
mammalian DNA ligases. Mutat. Res., 407, 1–9.
increases spontaneous mutation and alkylation sensitivity in Escherichia
15. Wilson,D.M. and Thompson,L.H. (1997) Life without DNA repair. Proc.
coli. J. Bacteriol., 181, 6763–6771.
Natl Acad. Sci. USA, 94, 12754–12757.
16. Tsurudome,Y., Hirano,T., Yamato,H., Tanaka,I., Sagai,M., Hirano,H.,
Received on July 26, 2000; revised on September 22, 2000; accepted on
September 25, 2000
Source: http://rusynlab.com/publications/2000/Rusyn%20et%20al%20Carcinogenesis%202000.pdf
AP® STATISTICS 2011 SCORING GUIDELINES (Form B) Question 2 Intent of Question The primary goals of this question were to assess students' ability to (1) distinguish an experiment from an observational study; (2) critique statistical information, in particular whether or not researchers are justified in making a specific conclusion based on the given information; (3) recognize and describe a potential problem with a study that lacks random assignment or blinding. Solution Part (a):
expand new drug markets for TB endTB aims to find shorter, less toxic and more effective treatments for multidrug-resistant TB 5 Approach through access to new drugs, a clinical trial, 6 Countries and advocacy at country and global levels. 7 Country requirements Only 11% of multidrug-resistant tuberculosis (MDR-TB) patients get


