
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Forschungsbericht_ebergeruch_fin2
Rheinische Friedrich-Wilhelms-Universität Bonn
Landwirtschaftliche Fakultät
Lehr- und Forschungsschwerpunkt
„Umweltverträgliche und Standortgerechte Landwirtschaft"
Molecular genetic analysis of boar taint
Verfasser:
Prof. Dr. Christian Looft
Institut für Tierwissenschaften
Professur für Tierzucht und Tierhaltung
Herausgeber: Lehr- und Forschungsschwerpunkt „Umweltverträgliche und Standort-gerechte
Landwirtschaft", Landwirtschaftliche Fakultät der Rheinischen Friedrich-Wilhelms-
Universität Bonn
Meckenheimer Allee 172 15, 53115 Bonn
Tel.: 0228 – 73 2285; Fax.: 0228 – 73 1776
www.usl.uni-bonn.de
Forschungsvorhaben im Auftrag des Ministeriums für Umwelt und
Naturschutz, Landwirtschaft und Verbraucherschutz des Landes
Nordrhein-Westfalen
Bonn, September 2012
Prof. Dr. Christian Looft
Projektbearbeitung: C. Neuhoff, A. Gunawan, L. Frieden, M. Pröll, Dr. C. Große-
Brinkhaus, Dr. M.U. Cinar, Prof. Dr. K. Schellander, Dr. E. Tholen
Institut für Tierwissenschaften
Professur für Tierzucht und Tierhaltung
Endenicher Allee, 53115 Bonn
Tel.: 0228/73 9328; Fax.: 0228/73 2284
Neuhoff C., A. Gunawan, M. Pröll, L. Frieden, C. Große-Brinkhaus, E. Tholen, M.U. Cinar,
K. Schellander, C. Looft (2012): Molecular genetic analysis of boar taint. Landwirtschaftliche
Fakultät der Universität Bonn, Schriftenreihe des Lehr- und Forschungsschwerpunktes USL,
Nr. 170, 48 Seiten
Table of contents
Problem/Knowledge
Material und Methods
Animals and phenotypes
DNA and RNA isolation
Gene expression analysis with Affymetrix chips
Gene expression analysis with RNA-Seq
SNP-genotyping for the association study and statistical analysis
Gene expression analysis with Affymetrix chips
Analysis of RNA-Seq data
Differential gene expression analysis based on RNA-Seq
Validation of selected DEGs with quantitative Real Time PCR (qRT-PCR)
Gene variation analysis
Association between candidate genes and boar taint compounds
Consequences for practical agriculture
Schlussfolgerungen für die Umsetzung der Ergebnisse in die Praxis
Consequences for further research
Venn diagram of differentially expressed genes in the different analysed
Heatmap showing differentially expressed genes in (A) testis and (B) liver
Functional grouping of DEGs in testis with high and low androstenone using
Ingenuity Pathways Analysis (IPA) software.
The most prominent canonical pathways related to the DEGs data (
p < 0.05)
for testis with high and low androstenone.
Functional grouping of DEGs in liver with high and low androstenone using
Ingenuity Pathways Analysis software.
Gene network showing the relationship between molecules differentially
expressed in high androstenone testis samples.
Gene network showing the relationship between molecules differentially
expressed in high androstenone liver samples.
qRT-PCR validation for fourteen DEGs from divergent androstenone levels in
(A) testis and (B) liver samples.
Details of primers used for qRT-PCR analysis
Polymerase chain reaction primers used for SNPs screening
Differentially expressed genes based on microarrays – high skatol group
versus low skatol group
Differentially expressed genes – high andostenone group versus low
androstenone group
Summary of sequence read alignments to reference genome in testis samples
Summary of sequence read alignments to reference genome in liver samples
Differentially expressed genes in testis androstenone samples
Differentially expressed genes in liver androstenone samples
Functional categories and corresponding DEGs in high androstenone testis
Functional categories and corresponding DEGs in high androstenone liver
Polymorphisms detected in testis samples
Polymorphisms detected in liver samples
Genotype frequencies for tested genes.
Genotype and association analysis of candidate genes and boar taint
1 Introduction
1.1 Problem/Knowledge
Intact boars are rarely used for fattening, because consumers would object to the boar taint,
which tends to develop with sexual maturity and renders pork inedible. To eliminate this
problem, boars are usually castrated at a young age, a practice which is painful and has been
criticized repeatedly as not in line with animal welfare. In 2008, representatives of the
German pig farming community, the processing industry and the trade drafted a resolution
(„Düsseldorfer Erklärung") to stop castration of piglets without anesthezation. European pig
farmers and their union (COPA-COGECA) agreed in December 2010 to terminate surgical
castration by 2018. This means that castration of piglets with anesthesia will only be accepted
as a transitional step until castration will be completely banned in Europe. However, if intact
boars are fattened, negative consumer response to boar taint in pork has to be prevented: by
testing carcasses routinely with sufficient speed and accuracy and by reducing the incidence
of boar taint at slaughter age. This may be approached in different ways: by genetic selection,
nutrition and/or management.
Boar taint develops under the influence of genetic and non-genetic factors (Bracher-Jakob,
2000). Several studies have shown that the level of skatole and androstenone, the two main
components responsible for boar taint, is moderately to highly heritable; the deposition in fat
increases with sexual maturity. Non-genetic contributing factors which have been identified
are group
vs. single pen management and light for
androstenone level and nutrition, housing
system and hygiene for
skatole.
In order to assess the chances to reduce and eventually eliminate the boar taint by genetic
selection, we need to know the relevant population parameters. These estimates should not be
taken at face value without taking all essential factors into account: age and live weight at the
time of testing, management conditions, laboratory techniques applied, and sample size. As
pointed out by Haugen (2010), neither are official reference methods available to determine
and compare androstenone and skatole levels, nor are all results being published.
The relevance of laboratory techniques has been demonstrated by Harlizius et al. (2008),
whose results from different laboratory methods differed by a factor of 2 to 4 for identical
samples of backfat. This should be kept in mind; for genetic evaluation, genotypes must
always be compared under the same conditions.
A number of quantitative trait loci (QTL) and genome-wide association analysis have been
conducted for androstenone in the purebred and crossbred pig populations (Duijvesteijn et al.,
2010; Gregersen et al., 2012; Grindflek et al., 2011; Lee et al., 2004; Quintanilla et al., 2003;
Robic et al., 2011). Gene expression analysis has been used to identify candidate genes related
to the trait of interest. Several candidate genes have been proposed for divergent androstenone
levels in different pig populations by global transcriptome analysis in boar testis and liver
samples (Leung et al., 2010; Moe et al., 2008; Moe et al., 2007). Functional genomics
provides an insight into the molecular processes underlying phenotypic differences
(Ponsuksili et al., 2011). RNA-Seq is a recently developed next generation sequencing
technology for transcriptome profiling that boosts identification of novel and low abundant
transcripts (Wang et al., 2009). RNA-Seq also provides evidence for identification of splicing
events, polymorphisms, and different family isoforms of transcripts (Marguerat and Bahler,
1.2 Objectives
The aim of this study was the identification of genes and pathways influencing boar taint and
involved in androstenone and skatol metabolism. Therefore polymorphisms in relevant genes
were identified and transcriptome analysis using Affymetrix-Chips and RNA-Seq in the two
major organs, testis and the liver, involved in androstenone and skatole metabolism was
2 Material und Methods
2.1 Material
2.1.1 Animals and phenotypes
Tissue samples and phenotypes were collected from the Pietrain × F2 cross and Duroc × F2
cross animals. F2 was created by crossing F1 animals (Leicoma × German Landrace) with
Large White pig breed. Fattening performances of each boar was determined on station for
116 days. Animals were slaughtered when on average 90 kg gain was achieved during this
test. All the pigs were slaughtered in a commercial abattoir. Carcass and meat quality data
were collected according to guidelines of the German performance test (ZDS, 2007). Tissue
samples from testis and liver were frozen in liquid nitrogen immediately after slaughter and
stored at -80°C until used for RNA extraction. Fat samples were collected from the neck and
stored at -20°C until used for androstenone measurements. For the quantification of
androstenone an in-house gas-chromatography/mass spectrometry (GC-MS) method was
applied as described previously (Fischer et al., 2011). Pigs having a fat androstenone level
less than 0.5 µg/g and greater than 1.0 µg/g were defined as low and high androstenone
samples, respectively.
2.1.2 DNA and RNA isolation
For the microarray study, 20 animals of 101 crossing boars (Pietran x F2) with high and low
androstenone and skatole levels were selected. Average levels of androstenone were at > 470
ng/g fat and of skatole at > 250 ng/g fat.
Based on next generation sequencing techniques ten boars (Duroc x P2) were investigated.
These were selected from a pool of 100 pigs and the average androstenone value for these
selected animals was 1.36 ± 0.45 µg/g. RNA for RNA-seq was isolated from testis and liver
of 5 pigs with extreme high (2.48 ± 0.56 µg/g) and 5 pigs with extreme low levels of
androstenone (0.24 ± 0.06 µg/g).
In general total RNA was extracted using RNeasy Mini Kit according to manufacturer's
recommendations (Qiagen). Total RNA was treated using on-column RNase-Free DNase set
(Promega) and quantified using spectrophotometer (NanoDrop, ND8000, Thermo Scientific).
RNA quality was assessed using an Agilent 2100 Bioanalyser and RNA Nano 6000 Labchip
kit (Agilent Technologies).
For further investigation, selected candidate genes were genotyped in 300 crossing boars
(Pietran x F2). Therefore DNA was obtained from muscle tissue using a phenol-chloroform
extraction method.
2.2 Methods
2.2.1 Gene expression analysis with Affymetrix chips
Liver gene expressions pattern were produced using 20 GeneChip Porcine Array
(Affymetrix). The analysis of microarray raw data was performed with the R software
(http://www.r-project.org). For normalization and background correction of the data, the
algorithm gcRMA (GeneChip Robust Multichip Average) was used. Carrying out the analysis
of expression differences was performed with a linear model for microarray data (limma)
(Smyth, 2004). Three comparisons were taken into account by means of linear contrasts: (1)
the comparison of high vs. low skatole, (2) high vs. low androstenone and (3) the interaction
between skatole and androstenone. Differentially regulated genes were identified on the basis
of a p ≤ 0.05, one fold changes ≥1 and a false discovery rate (FDR) ≤ 0.3. The functional
annotation of differentially expressed genes was performed by the DAVID (The Database for
2.2.2 Gene expression analysis with RNA-Seq
Library construction and sequencing
Full-length cDNA was obtained from 1 µg of RNA, with the SMART cDNA Library
Construction Kit (Clontech, USA), according to the manufacturer's instructions. Libraries of
amplified RNA for each sample were prepared following the Illumina mRNA-Seq protocol.
The library preparations were sequenced on an Illumina HiSeq 2000 as single-reads to 100 bp
using 1 lane per sample on the same flow-cell (first sequencing run) at GATC Biotech AG
(Konstanz, Germany). All sequences were analysed using the CASAVA v1.7 (Illumina,
Reference sequences and alignment
Two different reference sequence sets were generated from NCBI Sscrofa 9.2 assembly. (1)
The reference sequence set generated for differential expression analysis comprised of RefSeq
mRNA sequences (cDNA sequences) and candidate transcripts from NCBI UniGene database
(Sscrofa). (2) For gene variation analysis a different reference sequence set, generated from
whole genome sequence (chromosome assembly) was used. During sequencing experiment
Sscrofa NCBI 10.2 assembly was not released and Sscrofa 9.2 covered 8.5 K unannotated
SNPs (dbSNP database). The released Sscrofa 10.2 assembly consists of 566 K SNP
annotation information for 460 K SNP (dbSNP database). In order to make use of this
(http://www.ncbi.nlm.nih.gov/genome/tools/remap) to convert Sscrofa 10.2 SNP genomic
positions to Sscrofa9.2 positions. Raw reads were mapped to reference sets using BWA
algorithm (http://bio-bwa.sourceforge.net/) with the default parameters (Li and Durbin, 2009).
Differential gene expression analysis
For differential gene expression analysis with raw count data a R package DESeq was used
(Anders and Huber, 2010). To model the null distribution of the count data, DEseq follows an
error model that uses the negative binomial distribution, with variance and mean linked by
local regression. The method controls type-I error and provides good detection power (Anders
and Huber, 2010). After analysis using DESeq, DEGs were filtered based on
p-adjusted value
(Benjamini and Hochberg, 1995) 0.05 and fold change > 1.5.
Gene variation analysis
For gene variation analysis the mapping files generated by aligning the raw reads to reference
sequence set (2) were used. All the downstream analysis was performed using Genome
(http://picard.sourceforge.net/). The Genome Analysis Toolkit (GATK) was used for local
realignment incorporating Sscrofa 9.2 converted SNPs which was described in the previous
section. Covariate counting and base quality score recalibration were done using the default
parameters suggested by GATK toolkit. The re-aligned and recalibrated mapping files were
grouped according to tissue and phenotype categories. Variant calling was performed for each
group using GATK UnifiedGenotyper (McKenna et al., 2010). All the variant calls with a
read coverage depth < 75 and base quality < 20 were discarded from further analysis.
Polymorphisms identified in DEGs are given in the results section.
Pathways and networks analysis
A list of the DEGs was uploaded into the Ingenuity Pathway Analysis (IPA) software
(Ingenuity Systems, www.ingenuity.com) to identify relationships between the genes of
interest and to uncover common processes and pathways. Networks of the genes were then
algorithmically generated based on their connectivity. The ‘Functional Analysis' tool of the
IPA software was used to identify biological functions which were most significant to the data
set. Canonical pathway analysis was also utilized to identify the pathways from the IPA
library of canonical pathways that were most significant to the data set. Fisher's exact test was
used to calculate a
p-value determining the probability that each biological function or
canonical pathway assigned to the data set. In addition, the significance of the association
between the data set and the canonical pathway was calculated as the ratio of the number of
genes from the data set that mapped to the pathway divided by the total number of genes that
mapped to the canonical pathway.
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA from testis and liver was isolated from 10 boars for qRT-PCR experiment. cDNA
were synthesised by reverse transcription PCR using 2 µg of total RNA, SuperScript II
reverse transcriptase (Invitrogen) and oligo(dT)12 primer (Invitrogen). Gene specific primers
for the qRT-PCR were designed by using the Primer3 software (Rozen and Skaletsky, 2000).
Detailed information for primers used in this study was given in Table 1. Nine-fold serial
dilution of plasmids DNA was prepared and used as a template for the generation of the
standard curve. In each run, the 96-well microtiter plate contained each cDNA sample,
plasmid standards for the standard curves and no-template control. For each PCR reaction 10
µl iTaqTM SYBR® Green Supermix with Rox PCR core reagents (Bio-Rad), 2 µl of cDNA
(50 ng/µl) and an optimized amount of primers were mixed with ddH2O to a final reaction
volume of 20 µl per well. The qRT-PCR was conducted with the following program: 95 °C
for 3 min and 40 cycles 95 °C for 15 s/60 °C for 45 s on the StepOne Plus qPCR system
(Applied Biosystem). As a technical replication, all samples were repeated and the mean of
the two replications was finally used. Final results were reported as the relative expression
level compared after normalization of the transcript level using two housekeeping genes PPIA
Details of primers used for qRT-PCR analysis
Primer sequences (5'→3')
F: AGCTGTCGATGGAGCAAGTT
R: CCACATCCAAAGGCCTTAAA
F: GTTTGCATCTTGGGGACACT
R:ATGGGAACAGCTCTTGAGGA
F: AGCACCCTGAAGTCTCTGGA
R:GACAGGATGAGGAGGAGCTG
F: TGTTGAAGAGCCATGGACAA
R: CTTCAGCAGAGGGAAGTTGG
F:TCCTGATGACAAAGGCAGTG
R:TGCCTTATCCATCCACAACA
F: AGCTGTGCCTCATCCCTAGA
R: GTGTTTCTGTCCCAGGCAAT
F:GTGACGGAAGAAACCGTAA
R: CTCCAGGGACTCTGAACTGC
F:TCCCCAGTGTTTTCTGGTTC
R:CCTTCTCCTCCAGCAACAAG
F: TGCAGAACAGAGGACTGTGG
R: GCCATGCATCGTTTGTATTG
F: CCTGCCAGCGAGAACTCTAC
R: CTCGCACTGTTTGCTGTGAT
F: TTCCCGATTCATGTGTTCAA
R: ACCAGTTCCGAGATGTGGTC
F:ACTGGCTGGTAGGTCCCTTT
R:TCTCAGGTTGCTGGGTCTCT
F:GGCCTGAAGCCTAAACACAG
R:CCTGGAGCCATCCTCAAATA
F: CACAAACGGTTCCCAGTTT
R: TGTCCACAGTCAGCAATGGT
F:ACCCAGAAGACTGTGGATGG
R:ACGCCTGCTTCACCACCTTC
2.2.3 SNP-genotyping for the association study and statistical analysis
To identify polymorphisms within candidate genes, specific primers were designed based on
published sequences by using Primer3 software (Rozen and Skaletsky, 2000).
A list of primers used in this study is given in Table 2.
Polymerase chain reaction primers used for SNPs screening
Rv: 5´-TGTGCTGGTAATGGCACAAA-3´
Fw: 5´-AATTCTGCACATTCCCCTGA-3´
Rv: 5´-CCTGTTTGTTTCCTTGATTGC-3´
Fw: 5´- GTTCAAATCCCTGGTTGCAT-3´
Rv: 5´-CTAGGCGTCTCCCCAGATTAG-3´
Fw: 5´-GGTAACCTGTCCCCTCCTG-3´
Rv: 5´-GGTAAGAGACGGCACAGGAG-3´
Fw: 5´-TCAAGGCACTCAGGATAAGC-3´
Rv: 5´-GAACACTGAGGAGCCTGGTA-3´
Fw: 5´- TCAAGGCACTCAGGATAAGC-3´
Rv: 5´- GAACACTGAGGAGCCTGGTA-3´
Polymerase Chain Reactions (PCR)
Polymerase Chain Reactions were performed in a 20 µl volume containing 2 µl of genomic
DNA, 10×PCR buffer (with 2.0 µl MgCl2), 1.0 µl of dNTP, 0.5 µl of each primer and 0.2 µl
of Taq DNA polymerase (GeneCraft). The PCR were performed under the following
condition: initial denaturing at 95 ºC for 5 min followed by 35 cycles of 30 sec at 95 ºC, 30
sec at respective annealing temperatures (as given in Table 4) and 10 sec at 72 ºC and a final
elongation of 10 min at 72 ºC .
The PCR-RFLP method was used for genotyping the boars. The restriction enzymes were
selected according to the recognition (http://tools.neb.com/NEBcutter2/index.php) of the
polymorphic sites. The fragments with the detected mutation were amplified using different
annealing temperatures to get the the PCR products (Table 2).An aliquot of the PCR product
of each reaction was checked on 1.5% agarose gel (Fisher Scientific Ltd.) before digestion
using different endonucleases. The digested products were separated using 2.0% agarose gel.
The fragments were visualised under ultraviolet light, and the sizes and the number of
fragments analysed using the molecular analyst software (Bio-Rad Laboratories, Molecular
Bioscience Group).
Statistical Analysis of the association study
Allele and genotype frequencies of each population were determined to detect SNP in the six
candidate genes. The association of the genotypes from six candidate genes with boar taint
compounds were calculated by analyzing variance of quantitative traits. For these analyses a
generalized linear model of SAS (SAS Inst. Inc., Cary, NC) was used. The model was as
Y
ijklm = µ
+ season
i + genotype
j + station
k + pen
l + e
ijkl
Where Y is the boar taint compounds (Skatole, Androstenone and Indole), µ
is overall mean,
season
is the fixed effect of i-th season (i= winter/summer), genotype is the fixed effect of j-th
genotype (j=1,2, and 3), station is the fixed effect of k-th station (Grub, Schwarzenau,
Frankenforst, Haus Düsse and Boxberg), pen is the fixed effect of l-th pen (group, individual),
and e
ijkl is the residual error.
The distribution of the genotypes and accuracy of genotype scoring was tested for Hardy–
Weinberg equilibrium by chi-square (X2) test before using both polymorphisms for the
association analysis.
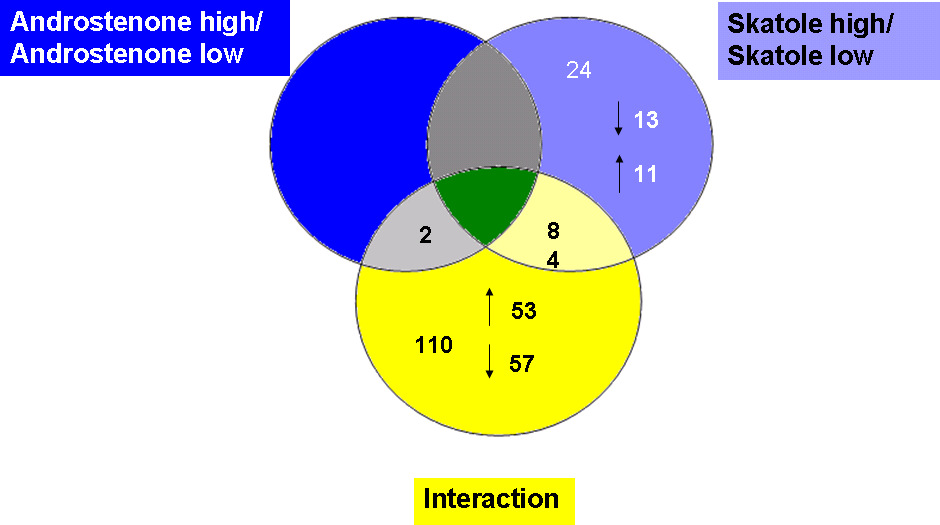
3 Results
3.1 Gene expression analysis with Affymetrix chips
Differentially regulated genes based on the comparison of high vs. low skatole and high vs.
low androstenone are described in Table 3 and Table 4. Generally 107 genes were
differentially expressed comparing high and low skatole. 49 were up regulated and 58 were
down regulated. The investigation of differentially expressed genes related to a divergent
andostrenone level revealed only two genes (Figure 1).
Venn diagram of differentially expressed genes in the different analysed groups
A gene ontology classification was performed using the online tool DAVID in order to assign
differentially expressed genes to categories biological functions and pathways. Differentially
expressed genes between the respective groups showed significant features in catalytic
activities, metabolic processes, fatty acid metabolism and lipid metabolic processes.
Investigating the data using an interaction term between skatole and andostenone revealed a
different set of differentially expressed genes. The gene FMO1 (Flavin containing
monooxygenase 1) was identified within this step, and seems to be promising, because it is
involved in the phase I metabolism of skatole and andostenone.
Differentially expressed genes based on microarrays – high skatol group versus
low skatol group
Gene symbol Gene name
cytochrome P450, family 4, subfamily A,
Acyl-CoA desaturase
Fatty acid synthase
Cytochrome P450 4A11
L-3-phosphoserine phosphatase
Lipid phosphate phosphohydrolase 1
Acetyl-coenzyme A synthetase, cytoplasmic
Dedicator of cytokinesis protein 1
Protein-tyrosine phosphatase delta precursor
Farnesyl pyrophosphate synthetase
tetratricopeptide repeat domain 21B
Aldehyde dehydrogenase 1A1
nei endonuclease VIII-like 1; endonuclease VIII
vacuolar protein sorting 13D
Delta(14)-sterol reductase
Acetyl-coenzyme A synthetase, cytoplasmic
Carbonic anhydrase VII
Lipid phosphate phosphohydrolase 1
similar to delta 5 fatty acid desaturase
7-dehydrocholesterol reductase
Acetyl-CoA carboxylase 1
Cytochrome P450 2D6
similar monocarboxylate transporter
Ankyrin 3 (ANK-3)
Protein-tyrosine phosphatase delta precursor
2-amino-3-ketobutyrate coenzyme A ligase,
mitochondrial precursor
heat shock-like protein 1
Angiotensinogen precursor
nei endonuclease VIII-like 1
UPF0143 protein C14orf1
UDP-glucuronosyltransferase 2B17 precursor,
Gene symbol Gene name
NAD(P)-dependent steroid dehydrogenase
Nuclear receptor ROR-alpha
HMG-BOX transcription factor BBX
Afamin precursor (Alpha-albumin)
Glutathione S-transferase theta 1
Complement-activating component of Ra-
reactive factor precursor
Aldehyde dehydrogenase 1A1
similar to delta 5 fatty acid desaturase
Solute carrier family 23, member 1
Valacyclovir hydrolase precursor
hyaluronan binding protein 2
Agmatinase, mitochondrial precursor
Short-chain dehydrogenase/reductase 3
Cytochrome P450 39A1
Tax1 binding protein
Group XIIA secretory phospholipase A2
Trifunctional enzyme alpha subunit,
mitochondrial precursor
Protein transport protein Sec23A
SNF-1 related kinase
PREDICTED: KIAA1423
Trans-Golgi network integral membrane protein
Integrin alpha-V precursor
Dolichyldiphosphatase 1
Protein CGI-100 precursor
Galactose-1-phosphate uridylyltransferase
Protein transport protein Sec23A
Isocitrate dehydrogenase [NADP] cytoplasmic
Mannose-6-phosphate receptor binding protein 1 1.273556
Brain protein 44.
ADAMTS-19 precursor
Gene symbol Gene name
Phosphoacetylglucosamine mutase
Adiponectin receptor protein 2
MARVEL domain containing 3;
Peroxisome proliferator activated receptor alpha
Mitochondrial carnitine/acylcarnitine carrier
Retinol dehydrogenase 11
Long-chain-fatty-acid--CoA ligase 1
Protein FAM34A. 4]
PR-domain zinc finger protein 6
Acyl-CoA dehydrogenase, very-long-chain
specific, mitochondrial precursor
Cell death activator CIDE-B (Cell death-
inducing DFFA-like effector B).
Glycerol-3-phosphate dehydrogenase [NAD+],
apoptosis regulator
Transmembrane 4 superfamily member 13
Ubiquinone biosynthesis monooxgenase COQ6
glycerol-3-phosphate dehydrogenase 1-like
I-mfa domain-containing protein isoform p40
I-mfa domain-containing protein isoform p40
Adiponectin receptor protein 2
ADP-ribosylation factor 4.
Platelet-activating factor acetylhydrolase
ATP-binding cassette, sub-family D, member 3
Dehydrogenase/reductase SDR family member 4 2.077268
ATP-binding cassette, sub-family D, member 3
L-lactate dehydrogenase B chain
ATP-binding cassette, sub-family D, member 3
Putative lymphocyte G0/G1 switch protein 2.
Gene symbol Gene name
Putative lymphocyte G0/G1 switch protein 2.
Phosphomannomutase 1
Hydroxymethylglutaryl-CoA synthase,
mitochondrial precursor
Differentially expressed genes – high andostenone group versus low
androstenone group
Gene symbol Gene name
log FC p-value FDR
Cytochrome P450 3A7
0.001722 0.844666
Inhibin beta A chain precursor
-1.06619 0.001891 0.844666
3.2 Analysis of RNA-Seq data
We sequenced cDNA libraries from 10 samples per tissue using Illumina HiSeq 2000. The
sequencing produced clusters of sequence reads with maximum 100 base-pair (bp) length.
After quality filtering the total number of reads for testis and liver samples ranged from 13.2
million (M) to 33.2 M and 12.1 M to 46.0 M, respectively. There was no significant
difference in the number of reads from low and high androstenone samples (p = 0.68). Total
number of reads for each tissue group and the number of reads mapped to reference sequences
are shown in Table 5 and Table 6. In case of testis 42.20% to 50.34% of total reads were
aligned to reference sequence whereas, in case of liver 40.8% to 56.63% were aligned.
Summary of sequence read alignments to reference genome in testis samples
Un-mapped Mapped
Sample number of
Low androstenone
High androstenone
Summary of sequence read alignments to reference genome in liver samples
Sample number of mapped
29,549,267 15,632,809 13,916,458 53.50
46,050,468 25,270,695 20,779,773 54.87
16,420,055 7,659,515
13,323,763 6,989,584
27,085,837 11,747,225 15,338,612 43.37
28,976,693 16,123,777 12,852,916 55.64
12,755,487 5,879,896
45,203,089 18,443,608 26,759,481 59.20
14,559,329 8,540,379
14,527,329 8,062,992
3.3 Differential gene expression analysis based on RNA-Seq
Differential gene expression for testis and liver with divergent androstenone levels were
calculated from the raw reads using the R package DESeq (Anders and Huber, 2010). The
significant scores were corrected for multiple testing using Benjamini-Hochberg correction.
We used a negative binomial distribution based method implemented in DESeq to identify
differentially expressed genes (DEGs) in testis and liver with divergent androstenone levels.
A total of 46 and 25 DEGs were selected from the differential expression analysis using the
criteria padjusted < 0.05 and fold change ≥ 1.5 for testis and liver tissues respectively (Table 7
and Table 8). In testis tissues, 14 genes were found to be highly expressed in high
androstenone group whereas, 32 genes were found to be highly expressed in low
androstenone group. In the liver tissue, 9 genes were found to be highly expressed in high
androstenone group whereas, 16 genes were found to be highly expressed in low
androstenone group (Table 7 and Table 8). The range of log fold change values for DEGs was
from -4.68 to 2.90 for testis and from -2.86 to 3.89 for liver. Heatmaps (Figure 1, A and B)
illustrate the DEGs identified in high and low androstenone testis and liver tissues. The
differential expression analysis of our data revealed both novel transcripts and common genes
which were previously identified in various gene expression studies. Novel transcripts from
our analysis and commonly found genes are mentioned in detail in the discussion section.
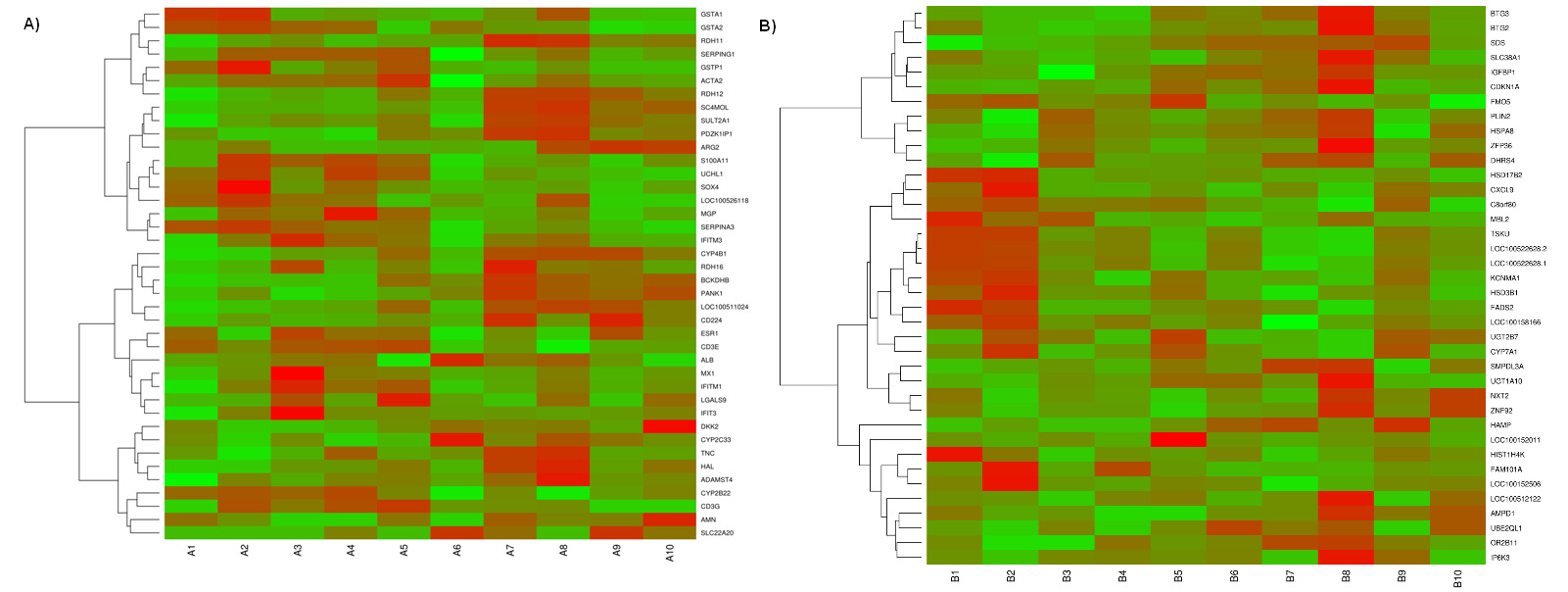
Heatmap showing differentially expressed genes in (A) testis and (B) liver
The red blocks represent over expressed genes, and the green blocks represent under
expressed genes. Legend: A1-A5 testis with low androstenone and A6-A10 testis with high
androstenone, B1-B5 liver with low androstenone and B6-B10 liver with high androstenone.
Differentially expressed genes in testis androstenone samples
p-adjusted
p-adjusted
Differentially expressed genes in liver androstenone samples
p-adjusted
To investigate gene functions and to uncover the common processes and pathways among the
selected DEGs, Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems,
www.ingenuity.com) was used. In testis samples, out of 46 DEGs 39 were assigned to a
specific functional group based on the information from IPA (Figure 3). A large proportion
(84.7%) of the DEGs from testis high androstenone group fell into Gene Ontology (GO)
categories such as molecular transport, small molecule biochemistry, amino acid metabolism,
embryonic development, carbohydrate metabolism, lipid metabolism and reproductive system
development and function (Figure 3).
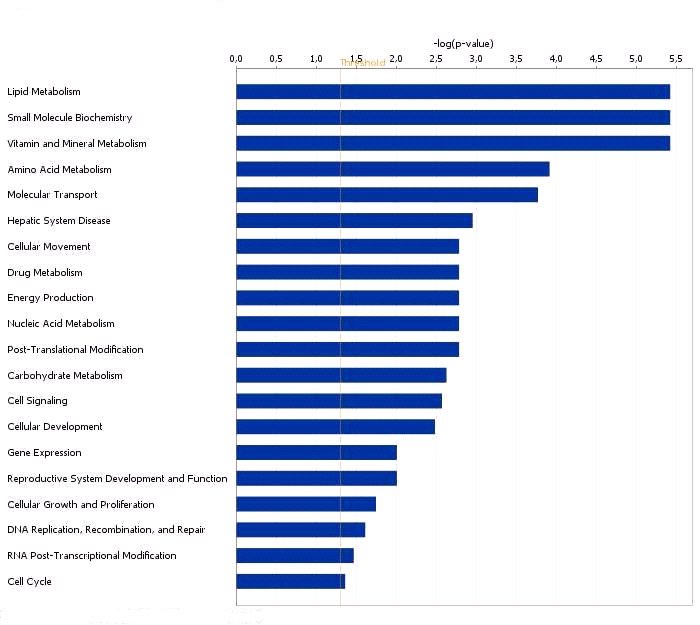
Functional grouping of DEGs in testis with high and low androstenone using
Ingenuity Pathways Analysis (IPA) software.
The most significant functional groups (p < 0.05) are presented graphically. The bars represent
the p-value on a logarithmic scale for each functional group.
The genes classified into each functional group are listed in the Table 9. The differentially
expressed genes MSMO1 and ARG2 are involved in arginine degradation metabolic pathway
and additionally, ARG2 is found to be involved in citruline biosynthesis and urea cycle
pathways. The gene MSMO1 is also involved in cholesterol biosynthesis and zymosterol
synthesis. The differentially expressed cytochrome family gene CYP4A11 is involved in
alpha-tocopherol degradation.
IPA assigned 104 DEGs between high and low androstenone testis samples to eleven different
canonical pathways. These enriched pathways were metabolic pathways including retinol,
trypthopan, arginine and proline, fatty acid and sulphur metabolism (Figure 4). Other pathway
categories, including LXR/RXR activation, valine, leucine & isolenone degradation,
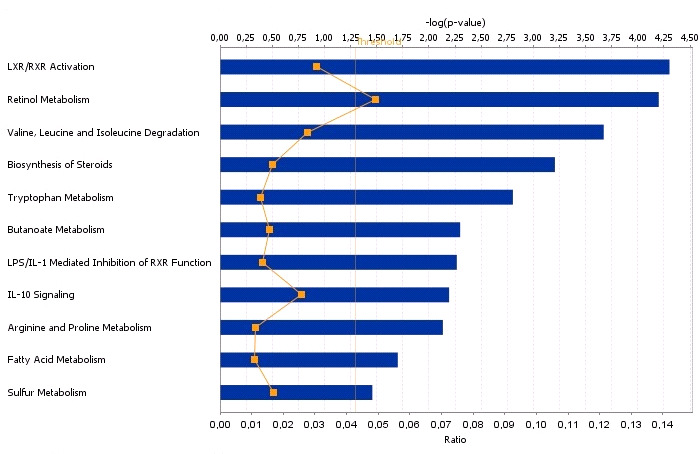
biosynthesis of steroid, butanoate, LPS/ILI mediated and IL-10 signaling were also enriched
The most prominent canonical pathways related to the DEGs data (p < 0.05) for
testis with high and low androstenone.
The bars represent the p-value for each pathway. The orange irregular line represents the ratio
(genes from the data set/total number of genes involved in the pathway) for the different
For the liver androstenone samples, out of 25 DEGs, 22 could be assigned to a specific
functional group based on the information from IPA (Figure 5). A large proportion (88.0%) of
the DEGs from liver high androstenone group was enriched with GO functional categories
such as amino acid metabolism, small molecule biochemistry, cellular development, lipid
metabolism, molecular transport, cellular function and maintenance and cellular growth and
proliferation. The genes classified into each group are listed in the Table 10. Among the
differentially expressed genes in liver samples, CDKN1A and HSD17B2 are involved in
VDR/RXR activation metabolic pathway and CYP7A1 and FMO5 genes are involved in
LPS/IL-1 mediated inhibition of RXR function pathway.
Functional categories and corresponding DEGs in high androstenone testis
HBB, HBD, HBA1/HBA2,
Molecular transport
1.00E-05 to 4.96E-02
CYP4A11,EDN1, MARCO, AMN,
1.00E-05 to 4.95E-02
HBA1/HBA2,CYP4B1, MX1, CYTL1,
CYP4A11, MARCO, MSMO1, DSP
Amino acid metabolism
3.80E-04 to 3.48E-02
ARG2, EDN1, HAL, FRK
Embrionic development
6.80E-04 to 4.40E-02
HBB, HBD, CYTL1, EDN1
Carbohydrate metabolism
7.54E-04 to 4.96E-02
CD244, EDN, CYTL1
CD244, EDN1,CYP4A11, HBB,
Lipid metabolism
7.54E-04 to 4.96E-02
MARCO, MSMO1, DSP
Reproductive system
1.95E-03 to 4.96E-02
development and function
Protein synthesis
1.03E-02 to 2.70E-02
HBA1/HBA2, HBB, ADAMTS4
Energy production
1.64E-03 to 2.43E-02
Vitamin and Mineral
1.50E-02 to 2.37E-02
EDN1, CD244, CD5
* Numbers in the p-value column showed a range of p-values for the genes from each category
IPA assigned 39 of DEGs in high and low androstenone liver group to 6 different canonical
pathways. Assigned canonical pathways were metabolic processes including retinol,
glycerolipid, fatty acid metabolism and xenobiotics metabolism by Cytochrome P450. Other
pathway categories, including PXR/RXR and VDR/RXR activation were also enriched.
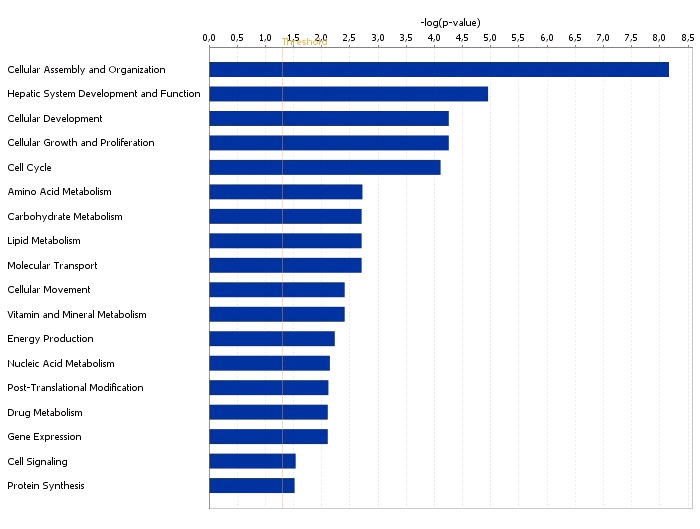
Functional grouping of DEGs in liver with high and low androstenone using
Ingenuity Pathways Analysis software.
The most significant functional groups (p < 0.05) are presented graphically. The bars represent
the p-value on a logarithmic scale for each functional group.
Functional categories and corresponding DEGs in high androstenone liver
Amino acid metabolism
8.71E-06 to 3.49E-02 HAL, SDS,CDKN1A,
HAL, CYP7A1, MBL2, AMPD3,
8.71E-06 to 2.51E-02
HSD17B2, IP6K1, SDS, CDKN1A
Cellular Development
3.15E-04 to 2.49E-02 CDKN1A, KRT8, HIST1H4A, MBL2
CYP7A1, MBL2, HSD17B2, IP6K1,
Lipid Metabolism
1.10E-03 to 2.41E-02 CDKN1A, KRT8
Molecular transport
1.11E-03 to 4.41E-02 CYP7A1, MBL2, CDKN1A
Cell Function and
1.20E-03 to 4.90E-02 CDKN1A, MBL2, KRT8, KRT18
1.20E-03 to 2.90E-02 CDKN1A, MBL2, KRT8
* Numbers in the p-value column showed a range of p-values for the genes from each category
In order to determine the biologically relevant networks other than canonical pathways,
network analysis was performed for DEGs in testis and liver samples. The networks describe
functional relationships between gene products based on known interactions reported in the
literature. Figure 6 exemplarily shows the network deduced from the list of functional
candidate genes from testis which are important for androstenone biosynthesis. The network
of testis androstenone level comprised of 16 focus genes belonging to functional categories
such as molecular transport, haematological disease and haematological system development
and function (Figure 6).
Gene network showing the relationship between molecules differentially
expressed in high androstenone testis samples.
Genes represented in this network are involved in lipid metabolism, small molecule
biochemistry and molecular transport. The network showed a relationship between genes
involved in the transport of lipid related molecules (ARL4C and CYP4A11) via blood system
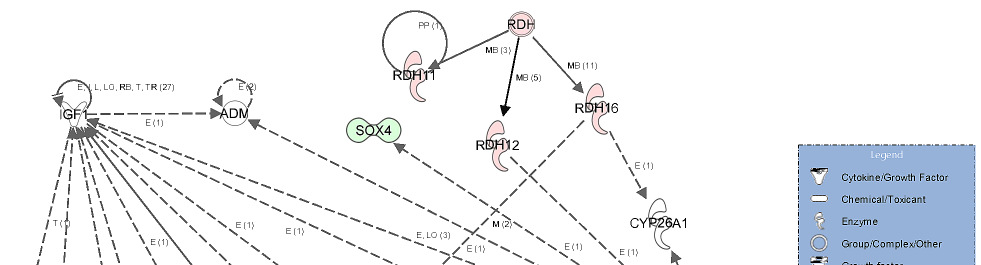
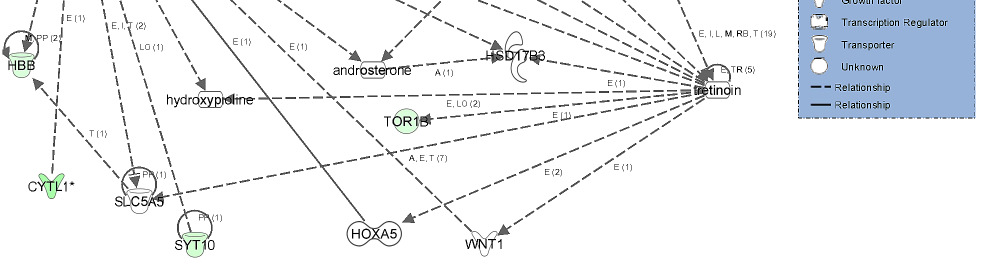
The second network of genes from liver androstenone contained 11 focus genes associated
with drug metabolism, endocrine system development and function and energy production
(Figure 7). The network shows the relationship between beta-estradiol and genes such as
FMO5, SMPDL3A and HSD17B2. The gene network shows that retinoid X receptor (RXR)
gene had direct relationship between PLIN2, CYP7A1 and NFkB genes and indirect
relationship with CDKN1A gene.
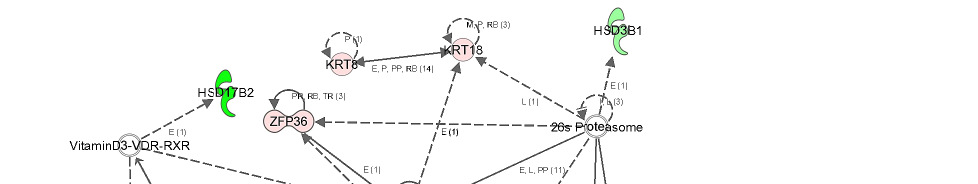
Gene network showing the relationship between molecules differentially
expressed in high androstenone liver samples.
Direct or indirect relationships between molecules are indicated by solid or dashed connecting
lines, respectively. The type of association between two molecules is represented as a letter on
the line that connects them. P, phosphorylation; A, gene activation; E, involved in expression;
PP, protein-protein interaction; PD, protein DNA-binding; MB, membership in complex; LO,
localization; L, proteolysis; RB, regulation of binding; T, transcription. The number in
parenthesis represents the number of bibliographic references currently available in the
Ingenuity Pathways Knowledge Base that support each one of the relationships. The intensity
of the color in the object is proportional to fold change.
3.4 Validation of selected DEGs with quantitative Real Time PCR (qRT-PCR)
In order to validate the RNA-Seq results, a total of 14 genes were randomly selected and
quantified using qRT-PCR. SULT2A1, DHRS4, ESR1, TNC, UCHL1, GSTA2 and CYP2C33
genes from testis samples and HSD3B1, CYP7A1, FMO5, IGFBP1, PLIN2, DHRS4 and
HSD17B2 genes from liver samples were selected for the validation by qRT-PCR.
Comparison of qRT-PCR data for 14 selected genes showed almost complete concordance of
expression with the RNA-Seq results (Figure 8, A and B).
qRT-PCR validation for fourteen DEGs from divergent androstenone levels in
(A) testis and (B) liver samples.
Fold change determined via division of high androstenone group gene expression value by low
androstenone group gene expression value. Gene expression values for qRT-PCR were
normalized using housekeeping genes PPIA and GAPDH.
3.5 Gene variation analysis
In total 222,225 and 202,249 potential polymorphism were identified in high and low
androstenone testis groups. Among these identified polymorphisms, 8,818 in high
androstenone group and 8,621 in low androstenone group were global polymorphisms with
reference and accession identifiers in dbSNP database. Similarly in liver high and low
androstenone samples 169,181 and 164,417 potential polymorphisms were identified. There
were 6,851 global polymorphisms in high androstenone liver sample and 6,436 global
polymorphisms in low androstenone liver sample.
Polymorphisms identified in DEGs for testis and liver samples are given in Table 11 and
Table 12. In the testis samples 12 gene polymorphisms were identified in 8 DEGs (Table 11).
Additionally our results revealed that mutations for the genes CD244 and ARG2 were specific
for high androstenone testis tissues, whereas mutations in genes IFIT2, DSP and IRG6 were
specific for low androstenone testis samples.
Thirty six mutations were identified in 11 DEGs in liver samples (Table 12). Variation in
HAL gene was specific for high androstenone liver samples whereas FMO5, HIST1H4K and
TSKU gene variations were specific for low androstenone liver samples (Table 12).
Polymorphisms detected in testis samples
High androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
High androstenone
High androstenone
High androstenone
Polymorphisms detected in liver samples
Alternate Quality
High Androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Low androstenone
Alternate Quality
Low androstenone
High Androstenone
High Androstenone
Low androstenone
High Androstenone
Low androstenone
3.6 Association between candidate genes and boar taint compounds
The distribution of genotype and allele in all six candidate genes is shown in Table 13. The
observed genotype frequencies for FMO1, CYP21, ESR1 and PLIN2 genes differed from
those expected from Hardy-Weinberg Law. In case of FMO5 and PLIN22 genes, the observed
genotype frequencies were according to expected values from Hardy-Weinberg Law.
Genotype frequencies for tested genes.
n.s=non-significant, χ ² =Chi-square test, p-value=deviation from Hardy-Weinberg Law
This study could not observe association of FMO1, PLIN2 and PLIN22 genotypes with boar
taint compounds in the population (Table 14).The result of association analysis of FMO5 gene
revealed significant association of additive effect and dominance effect with androstenone and
skatole respectively. The association analysis result of CYP21 revealed that there were
additive effects which involved with varying levels of skatole and indole respectively.
The association analysis of FMO5 gene revealed that animals with homozygote genotype
"GG" (6.07) had significantly increased androstenone level, whereas animals with
heterozygote genotype "AG" (4.88 and 3.93, respectively) had significantly increased skatole
and indole level .The association analysis of CYP21 gene revealed that animals with
homozygote genotype "CC" (5.13 and 4.27, respectively) had significantly increased skatole
and indole levels whereas in case of ESR1, the result of association analysis revealed that
animals with homozygote genotype "TT" and heterozygote genotype "CT" (6.15 and 4.36,
respectively) had significantly increased level of androstenone and indole respectively.
Genotype and association analysis of candidate genes and boar taint compounds.
Boar taint compound
Genotype (µ ± S.E.)
Effect (µ ± S.E.)
FMO1 g.256 A>C
Log Androstenone
FMO5 g.494 A>G
Log Androstenone
CYP21 g.3911 T>C
Log Androstenone
ESR1 g.672 C>T
Log Androstenone
Boar taint compound
Genotype (µ ± S.E.)
Effect (µ ± S.E.)
PLIN2 g.98 A>G
Log Androstenone
PLIN22 g.198 A>G
Log Androstenone
a, b,c * : P < 0.05, d,e,f ** : P < 0.001, Ln=natural log
4 Discussion
This study showed whole genome expression differences for varying androstenone levels in
testis and liver tissues. RNA-Seq provided high resolution map of transcriptional activities
and genetic polymorphisms in these tissues. However, due to incomplete porcine annotations,
only around 50% of the total reads could be mapped to annotated references. The
improvements in pig genome annotations may lead to better coverage and detailed
understanding of genetic and functional variants such as novel transcripts, isoforms, sequence
polymorphisms and non-coding RNAs. Integration of high throughput genomic and genetic
data (eQTL) with proteomic and metabolomic data can provide additional new insight into
common biological processes and interaction networks responsible for boar taint related traits.
On the basis of number of DEGs, our results confirm that transcriptome activity in testis is
higher in comparison to liver tissue for androstenone biosynthesis. These results also show
that the entire functional pathway involved in androstenone metabolism is not completely
understood and through this study, we propose additional functional candidate genes such as
SLC22A20, DKK2 and AMN in testis and HAMP, LOC100512122 and AADAT in liver.
Furthermore, various gene polymorphisms were also detected in testis and liver DEGs.
Potential polymorphisms were identified in DEGs such as HSP40, RASL11A and PDZK1IP1
in testis and PLIN2, IGFBP1, CYP7A1 and FMO5 in liver. These polymorphisms may have
an impact on the gene activity ultimately leading to androstenone variation and could be used
as biomarkers for boar taint related traits. Additionally, these potential biomarkers can also be
targeted for fertility and reproduction traits while breeding for boar taint. However, further
validation is required to confirm the effect of these biomarkers in other animal populations.
Furthermore this study revealed some significant results regarding the reduction of boar taint
and enhancing the fertility of boars which is the key question raised by animal breeders and
economists. It is not only important to cope up with problem of boar taint but this is equally
important that genes treating with boar taint should not affect the reproduction in boars.
Gunawan et al. (2011) reported the association of similar SNP of ESR1 with high sperm
quality and fertility traits. This aspect revealed the significance of this SNP as far as boar taint
and fertility in boars is concerned.
5 Summary
Boar taint is an unpleasant smell and taste of pork meat derived from some entire male pigs.
The main causes of boar taint are the two compounds androstenone (5α-androst-16-en-3-one)
and skatole (3-methylindole). It is crucial to understand the genetic mechanism of boar taint to
select pigs for lower androstenone levels and thus reduce boar taint. The aim of the present
study was to investigate transcriptome differences in boar testis and liver tissues with
divergent androstenone levels using microarrays and RNA deep sequencing (RNA-Seq).
The total number of reads produced for each testis and liver sample ranged from 13,221,550
to 33,206,723 and 12,755,487 to 46,050,468, respectively. In testis samples 46 genes were
differentially regulated whereas 25 genes showed differential expression in the liver. The fold
change values ranged from -4.68 to 2.90 in testis samples and -2.86 to 3.89 in liver samples.
Differentially regulated genes in high androstenone testis and liver samples were involved in
metabolic processes such as lipid metabolism, small molecule biochemistry and molecular
This study provides evidence for transcriptome profile and gene polymorphisms of boars with
divergent androstenone level using RNA-Seq technology. Digital gene expression analysis
identified candidate genes in flavin monooxygenease family, cytochrome P450 family and
hydroxysteroid dehydrogenase family. Moreover, gene polymorphism analysis revealed
potential mutations in IRG2, DSP, IFIT2, CYP7A1, FMO5 and CDKN1A genes in both high
and low androstenone sample groups. Further studies are required for proving the role of
candidate genes to be used in genomic selection against boar taint in pig breeding programs.
Additionally six genes FMO1, FMO5, CYP21, ESR1, PLIN2 and PLIN22 were selected for
association analysis based on their known function and their differential expression for boar
taint compounds. For the association studies, the SNP of six genes were genotyped in a total
of 370 animals. Three genes (FMO5, CYP21 and ESR1) were associated with boar taint
compounds. In detail, the association analysis of FMO5 showed its significant association
with all three boar taint compounds i.e., androstenone, skatole and indole whereas, ESR1
association results showed the association with androstenone and indole. According to the
results of association studies, FMO5, CYP21 and ESR1 turned out to be the most promising
candidates for boar taint.
6 Zusammenfassung
Ebergeruch ist eine unangenehme Geruchs- und Geschmacksabweichung im Schweinefleisch
von Ebern. Ebergeruch wird hauptsächlich durch die Stoffe Androstenon (5α-androst-16-en-
3-one) and Skatol (3-methylindole) hervorgerufen. Für die Selektion von Schweinen
bezüglich eines geringeren Androstenon- und Skatolgehalts, sowie einer damit verbundenen
geringeren Häufigkeit von Geruchsabweichungen, ist es notwendig, die grundlegenden
genetischen Mechanismen zu identifizieren. Das Ziel dieser Studie war es, Transkriptom-
Differenzen im Testis- und Leber-Gewebe von Tieren mit einem unterschiedlichen
Androstenon-Gehalten anhand von Microarray-Chips und der RNA-Sequenzierung (RNA-
Seq) zu untersuchen. Insgesamt 13,221,550 und 33,206,723 Sequenzen wurden für die
Testis-Proben generiert sowie 12,755,487 und 46,050,468 für die Leber-Proben.
Differentiell reguliert waren im Testis-Gewebe 46 Gene und im Leber-Gewebe 25 Gene. Die
„fold change"-Werte variierten zwischen -4.68 und 2.90 in den Testis-Proben und zwischen
-2.86 to 3.89 in den Leber-Proben. Die differentiell regulierten Gene aus der „Hoch-
Androstenon-Gruppe" waren an den metabolischen Prozessen Fettstoffwechsel, Biochemie
kleiner Moleküle und molekularer Transport beteiligt. Anhand der RNA-Sequenzierung
wurden in dieser Studie Transkriptom-Profile und Polymorphismen von Ebern mit deutlich
unterschiedlichen Androstenon-Gehalten dargestellt. Die Genexpressionsanalyse identifizierte
die Kandidatengene in den flavin monooxygenease, cytochrome P450 und hydroxysteroid
Polymorphismus-Analyse
Mutationen in den Genen IRG2, DSP, IFIT2, CYP7A1, FMO5 und CDKN1A sowohl in der
hohen als auch in der niedrigen Androstenon Gruppe. Weitere Studien sind notwendig, um die
Bedeutung der Kandidaten-Gene zu analysieren, bevor diese für die Genomische Selektion
gegen Ebergeruch in Zuchtprogrammen genutzt werden können.
Auf Grund ihrer Funktion und ihrer differentiellen Expression wurden die Gene FMO1,
FMO5, CYP21, ESR1, PLIN2 and PLIN22 für Assoziations-Studien ausgewählt. 370 Tiere
wurden für SNPs dieser Gene genotypisiert. Die Gene FMO5, CYP21 und ESR1 zeigten
Assoziationen zu den Ebergeruchs Merkmalen, wobei FMO5 signifikante Assoziationen zu
Androstenon, Skatol und Indol zeigte. ESR1 war mit Androstenon und Indol assoziiert. Die
Assoziationsstudie zeigte, dass FMO5, CYP21 and ESR1 vielversprechende Kandidatengene
für Ebergeruchsmerkmale sind.
7 Consequences for practical agriculture
It is obvious that castration of piglets with anesthesia will only be accepted as a transitional
step until castration will be completely banned in Europe. However, if intact boars are
fattened, negative consumer response to boar taint in pork has to be prevented: by testing
carcasses routinely with sufficient speed and accuracy and by reducing the incidence of boar
taint at slaughter age. This may be approached in different ways: by genetic selection,
nutrition and/or management.
On first sight, genomic selection may seem to offer a quick and easy solution. Before drawing
premature conclusions, the results of Grindflek et al. (2010) should be noted who found
markers for fertility traits on the same locations of the chromosome as for androstenone level,
which is not surprising in view of the described antagonistic effects. Moreover associations
between markers and traits are known to be breed specific. In any case, genetic markers have
to be identified in each population, with relevant correlations to other traits, before genomic
selection is applied in practice.
The intensity of boar taint in carcasses of intact boars can be reduced by selection. This can
help the pork industry in gradually reducing the number of carcasses discarded because of
boar taint and eventually eliminate the need for castration. To achieve optimal response to
selection, standardized procedures for measuring the two main components of boar taint,
androstenone and skatole, should be developed. Two current research projects (Anon.,
2009a,b) are focused on the development of automated measurement of boar taint for use in
processing plants as well as on live animals. The eventual goal is to develop techniques for
screening live boars for taint score, based on microbiopsy of backfat, saliva or blood samples,
which would speed up genetic progress.
The rate at which genetic progress can be reached will depend on antagonistic correlation
between boar taint and reproductive traits. These genetic correlations have to be determined in
relevant commercial male and female lines.
When identified QTLs for boar taint are being used in genomic selection, special attention
should be on gene locations which are not known to be negatively correlated with
reproductive performance.
8 Schlussfolgerungen für die Umsetzung der Ergebnisse in die Praxis
Grundsätzlich lässt sich der Anteil genussuntauglicher Eberschlachtkörper züchterisch
reduzieren. Voraussetzung hierfür ist jedoch, dass die Erfassung der beiden Leitkomponenten
Skatol und Androstenon standardisiert ist und damit eine laborübergreifende Vergleichbarkeit
ermöglicht wird. Derzeit werden im Rahmen von zwei Forschungsprojekten (Anon, 2009a,b)
die Möglichkeiten einer automatisierten Erfassung des Ebergeruchs für züchterische Zwecke
und zur Sortierung im Schlachtprozess untersucht. Die Entwicklung von Technologien zur
routinemäßigen Erfassung des Ebergeruchs am lebenden Zuchteber mit Hilfe von
Mikrobiopsie-, Speichel- oder Blutproben wären im Sinne schneller Zuchterfolge
Der Erfolg entsprechender Zuchtprogramme wird in entscheidender Weise durch das Ausmaß
der zu erwartenden antagonistischen Beziehungen zwischen Reproduktionsmerkmalen und
Ebergeruch beeinflusst. Entsprechende populationsspezifische Untersuchungen sollten
durchgeführt werden, um die Vereinbarkeit beider Selektionsziele beurteilen zu können.
Durch die Berücksichtigung identifizierter QTL im Rahmen der Genomischen Selektion ist
eine Steigerung der Selektionserfolge zu erwarten. Besonderes Augenmerk ist dabei auf
Genorte zu legen, mit deren Hilfe die gegenläufige Beziehung der beiden Merkmalskomplexe
Fruchtbarkeit und Ebergeruch aufgebrochen werden kann.
9 Consequences for further research
Results concerning the functional pathway involved in androstenone and skatole metabolism
will be integrated into the project STRAT-E-GER, Strategien zur Vermeidung von
Geruchsabweichungen bei der Mast unkastrierter männlicher Schweine (Fattening entire male
pigs - Strategies to prevent boar taint compounds), funded by the Bundesministerium für
Ernährung Landwirtschaft und Verbraucherschutz (BMELV), within the programme
Innovationsförderung. Association studies may confirm the biological significance of the
10 Patents
11 Publications
Neuhoff C, Pröll M, C. Große-Brinkhaus, L. Frieden, A. Becker, A. Zimmer, M.U. Cinar, E.
Tholen, C. Looft, K. Schellander (2011): Identifizierung von relevanten Genen des
Metabolismus von Androstenon und Skatol in der Leber von Jungebern mit Hilfe
von Transkriptionsanalysen. Vortragstagung der Deutschen Gesellschaft für
Züchtungskunde e.V. (DGfZ) und der Gesellschaft für Tierzuchtwissenschaften e.V.
(GfT), 6/7.9.2011, Freising-Weihenstephan, Deutschland
Frieden, L., Neuhoff, C., Große-Brinkhaus, Cinar, M.U., Schellander, K., Looft, C., Tholen, E
(2012): Züchterische Möglichkeiten der Verminderung der Ebergeruchsproblematik
bei Schlachtschweinen. Züchtungskunde, 84, 394-411
Gunawan, A., Sahadevan S. , Neuhoff, C., Große-Brinkhaus, C., Tesfaye, D., Tholen, E.
Looft, C., Schellander, K., Cinar, M.U. (2012): Using RNA-Seq for transcriptome
profiling in liver of boar with divergent skatole levels, P2035, ISAG meeting, Cairns,
Australien, 15.7.-20.7.2012
Neuhoff, C., Pröll, M., Große-Brinkhaus, C., Frieden, L., Becker, A., Zimmer, A., Tholen, E.,
Looft, C., Schellander, K. and Cinar, M.U. (2102): Global gene expression analysis
of liver for androstenone and skatole production in the young boars. p. 274, EAAP
meeting, Bratislava, Slovakia, 27.8.-31.8.2012
Gunawan, A., Sahadevan S. , Neuhoff, C., Große-Brinkhaus, C., Tesfaye, D., Tholen, E.
Looft, C., Schellander, K., Cinar, M.U. (2012): RNA deep sequencing analysis for
divergent androstenone levels in Duroc × F2 boars. Vortragstagung der Deutschen
Gesellschaft für Züchtungskunde e.V. (DGfZ) und der Gesellschaft für
Tierzuchtwissenschaften e.V. (GfT), 12/13.9.2012, Halle a.d. Saale, Deutschland
Gunawan, A., Sahadevan S. , Neuhoff, C., Große-Brinkhaus, C., Tesfaye, D., Tholen, E.
Looft, C., Schellander, K., Cinar, M.U. (2012): RNA deep sequencing reveals novel
candidate genes and polymorphisms in boar testis and liver tissues with divergent
androstenone levels, BMC Genomics, submitted
12 Presentations
Neuhoff C. (2011): Identifizierung von relevanten Genen des Metabolismus von Androstenon
und Skatol in der Leber von Jungebern mit Hilfe von Transkriptionsanalysen.
Vortragstagung der Deutschen Gesellschaft für Züchtungskunde e.V. (DGfZ) und der
Gesellschaft für Tierzuchtwissenschaften e.V. (GfT), 6/7.9.2011, Freising-
Weihenstephan, Deutschland
Neuhoff, C. (2012): Global gene expression analysis of liver for androstenone and skatole
production in the young boars. p. 274, EAAP meeting, Bratislava, Slovakia, 27.8.-
Gunawan, A. (2012): RNA deep sequencing analysis for divergent androstenone levels in
Duroc × F2 boars. Vortragstagung der Deutschen Gesellschaft für Züchtungskunde
e.V. (DGfZ) und der Gesellschaft für Tierzuchtwissenschaften e.V. (GfT),
12/13.9.2012, Halle a.d. Saale, Deutschland
13 Abstract
Boar taint is an unpleasant smell and taste of pork meat derived from some entire male pigs.
The main causes of boar taint are the two compounds androstenone (5α-androst-16-en-3-one)
and skatole (3-methylindole). It is crucial to understand the genetic mechanism of boar taint to
select pigs for lower androstenone levels and thus reduce boar taint. The aim of this study was
the identification of genes and pathways influencing boar taint and involved in androstenone
and skatol metabolism. Therefore polymorphisms in relevant genes were identified and
transcriptome analysis using Affymetrix-Chips and RNA-Seq in the two major organs
involved in androstenone metabolism i.e the testis and the liver was performed.
Differentially regulated genes in high androstenone testis and liver samples were involved in
metabolic processes such as retinol metabolism, metabolism of xenobiotics by cytochrome
P450 and fatty acid metabolism. Moreover, a number of genes encoding biosynthesis of
steroids were highly expressed in high androstenone testis samples. Gene polymorphism
analysis revealed potential mutations in HSP40, IGFBP1, CYP7A1 and FMO5 genes affecting
androstenone levels. Further studies are required for verify the role of candidate genes to be
used in genomic selection against boar taint in pig breeding programs. According to the
results of association studies, FMO5, CYP21 and ESR1 turned out to be the most promising
candidates for boar taint.
14 References
Anders, S., and W. Huber. 2010. Differential expression analysis for sequence count data.
Genome Biol 11: R106.
Benjamini, Y., and Y. Hochberg. 1995. Controlling the false discovery rate: A practical and
powerful approach to multiple testing. J R Stat Soc Series B 57: 289-300.
Bracher-Jakob, A. 2000. Jungebermast in forschung und praxis. Arbeit im Auftrag der
Eidgenössischen Forschungsanstalt für Nutztiere, 1725 Posieux, unter Leitung von
Duijvesteijn, N. et al. 2010. A genome-wide association study on androstenone levels in pigs
reveals a cluster of candidate genes on chromosome 6. Bmc Genetics 11.
Fischer, J. et al. 2011. Development of a candidate reference method for the simultaneous
quantitation of the boar taint compounds androstenone, 3alpha-androstenol, 3beta-
androstenol, skatole, and indole in pig fat by means of stable isotope dilution analysis
headspace solid-phase microextraction gas chromatography/mass spectrometry.
analytical chemistry 83: 6785-6791.
Gregersen, V. R. et al. 2012. Genome-wide association scan and phased haplotype
construction for quantitative trait loci affecting boar taint in three pig breeds. BMC
Genomics 13: 22.
Grindflek, E., I. Berget, M. Moe, P. Oeth, and S. Lien. 2010. Transcript profiling of candidate
genes in testis of pigs exhibiting large differences in androstenone levels. BMC
Grindflek, E. et al. 2011. Large scale genome-wide association and ldla mapping study
identifies qtls for boar taint and related sex steroids. BMC Genomics 12.
Gunawan, A. et al. 2011. Association study and expression analysis of porcine esr1 as a
candidate gene for boar fertility and sperm quality. Anim Reprod Sci 128: 11-21.
Harlizius, B. et al. 2008. Breeding against boar taint. An integrated approach. In: EAAP, 26.-
27. March - Girona
Haugen, J. E. 2010. Methods to detect boar taint. State of the art and critical review. EC
Workshop on pig welfare - castration of piglets, 2. June 2010, Brussels.
Lee, G. J. et al. 2004. Detection of quantitative trait loci for androstenone, skatole and boar
taint in a cross between large white and meishan pigs. Animal Genetics 36: 14-22.
Leung, M. C., K. L. Bowley, and E. J. Squires. 2010. Examination of testicular gene
expression patterns in yorkshire pigs with high and low levels of boar taint. Anim.
Biotechnol. 21: 77-87.
Li, H., and R. Durbin. 2009. Fast and accurate short read alignment with burrows-wheeler
transform. Bioinformatics 25: 1754-1760.
Marguerat, S., and J. Bahler. 2010. Rna-seq: From technology to biology. Cell Mol Life Sci
McKenna, A. et al. 2010. The genome analysis toolkit: A mapreduce framework for analyzing
next-generation DNA sequencing data. Genome Res 20: 1297-1303.
Moe, M. et al. 2008. Gene expression profiles in liver of pigs with extreme high and low
levels of androstenone. BMC Vet. Res. 4: 29.
Moe, M. et al. 2007. Gene expression profiles in testis of pigs with extreme high and low
levels of androstenone. BMC Genomics 8: 405.
Ponsuksili, S., E. Murani, B. Brand, M. Schwerin, and K. Wimmers. 2011. Integrating
expression profiling and whole-genome association for dissection of fat traits in a
porcine model. J Lipid Res 52: 668-678.
Quintanilla, R. et al. 2003. Detection of quantitative trait loci for fat androstenone levels in
pigs. Journal of Animal Science 81: 385-394.
Robic, A., G. Le Mignon, K. Feve, C. Larzul, and J. Riquet. 2011. New investigations around
cyp11a1 and its possible involvement in an androstenone qtl characterised in large
white pigs. Genetics Selection Evolution 43.
Rozen, S., and H. Skaletsky. 2000. Primer3 on the www for general users and for biologist
programmers. Methods Mol Biol 132: 365-386.
Smyth, G. K. 2004. Linear models and empirical bayes methods for assessing differential
expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3: article 3.
Wang, Z., M. Gerstein, and M. Snyder. 2009. Rna-seq: A revolutionary tool for
transcriptomics. Nat Rev Genet 10: 57-63.
Zentralverband der Deutschen Schweineproduktion (ZDS). 2007. Richtlinie für die
stationsprüfung auf mastleistung, schlachtkörperwert und fleischbeschaffenheit beim
schwein. Ausschuss für leistungsprüfung und zuchtwertschätzung (alz) beim schwein
des zentralverbandes der deutschen schweineproduktion (zds). Deutschland.
Source: https://www.usl.uni-bonn.de/pdf/forschungsbericht-170.pdf
Emergency Planning for Oyster Creek Important Safety Information For Your Community Please read the entire brochure or have someone translate it for you. Discuss this information with members of your family, and then keep the brochure in a convenient place for future use. ESTA INFORMACIÓN ES IMPORTANTE
This is a preprint version of the following article: Brey, P. (2008). ‘Human Enhancement and Personal Identity', Ed. Berg Olsen, J., Selinger, E., Riis, S., New Waves in Philosophy of Technology. New Waves in Philosophy Series, New York: Palgrave Macmillan, 169-185. Human Enhancement and Personal Identity 1. Introduction Human enhancement, also called human augmentation, is an emerging field within


