
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Whitesci.co.za
Mutagenesis Application Guide
Experimental Overview, Protocol, Troubleshooting
Mutagenesis Application Guide
Experimental Overview, Protocol, Troubleshooting
Senior Managing Editor and Contributor
Adam Clore, PhD, Brian Reinertson,
and Scott Rose, PhD
2011 Integrated DNA Technologies
Mutagenesis Application Guide
Mutagenesis Application Guide
Experimental Overview, Protocol, Troubleshooting
1.1 Site-directed Mutagenesis. . . . . . . . . . . . . . . 6
1.1.1 PCR for Substitutions, Additions, and Deletions . . . . . . . 7
1.1.4 Cassette Mutagenesis. . . . . . . . . . . . . . 16
1.2 Random Mutagenesis (in vitro saturation) . . . . . . . . . . 16
1.2.2 PCR with Degenerate Primers . . . . . . . . . . . . 19
1.2.3 Chemical Mutagenesis . . . . . . . . . . . . . . 19
2. Mutagenesis with Ultramer™ Oligonucleotides . . . . . . . . . 23
2.1.2 Terminal Additions by PCR . . . . . . . . . . . . 27
2.1.3 Oligonucleotide-directed Internal Mutagenesis . . . . . . 28
2.2.3 DpnI Digestion Controls . . . . . . . . . . . . . 30
2.3.1 Protocol for Terminal Changes or Additions . . . . . . . 31
2.3.2 Protocol for Oligonucleotide-directed Internal Mutagenesis . . 35
3.2 Template Concentration and Quality . . . . . . . . . . . 40
3.2.1 Too Much or Too Little Template . . . . . . . . . . . 40
3.5.5 Initial Denaturation Time. . . . . . . . . . . . . 44
3.6.1 Quantification of Product . . . . . . . . . . . . . 44
3.6.2 Gel Confirmation of a Ligation Reaction . . . . . . . . 45
3.8.1 Handling Competent Cells. . . . . . . . . . . . . 48
3.8.2 Heat Shock Considerations . . . . . . . . . . . . 48
3.8.3 Electroporation Considerations . . . . . . . . . . . 48
Mutagenesis Application Guide
1. Introduction
In vitro mutagenesis is used to purposefully change genetic information. Analysis of the subsequent changes in gene expression and gene products helps elucidate the func-tional effect of the mutation [1]. The technique falls into two general categories: site-directed mutagenesis and random mutagenesis. Each contains several subcategories with various methodologies used to generate mutations.
The particular mutagenesis method you choose to use will depend on the goal of the project and the information you have about the target sequence (Table 1). Site-directed mutagenesis creates a specific change in a known sequence, while random mutagen-esis allows researchers to screen for mutations regardless of the genomic location or to quickly create a wide variety of individual mutations. For both methods, a successful experiment depends on many factors including the technique used for constructing the mutant DNA, the quality of the primers used, an appropriate expression vector and system, effective purification, and development of an assay for detection [2].
This guide gives an overview of in vitro mutagenesis, assuming a pre-existing under-standing of standard cloning and PCR techniques. It also describes how long oligonucle-otides, called Ultramer™ Oligonucleotides, can simplify mutagenesis experiments. Two protocols for general site-directed mutagenesis techniques are provided. IDT supplies a variety of high quality reagents geared toward these methods (see IDT Reagents for Mutagenesis, pages 20 and 21). Many protocols and kits that simplify the mutagenesis process are also available commercially.
Mutagenesis Application Guide
If you are using a cloned
If you are using a genomic or
template source
cDNA template source
Limited base identity
PCR (Section 1.1.1)
PCR (Section 1.1.1)
changes at the end of the desired sequence
IDT reagent
25 nmole desalted oligos
25 nmole desalted oligos
Internal limited base identity Primer Extension
Primer Extension
changes (non-random)
IDT reagent
25 nmole desalted oligos or
25 nmole desalted oligos
Random internal base changes Error-prone PCR (Section 1.2.1) Error-prone PCR (Section 1.2.1)
5' or 3' terminal insertions
PCR (Section 1.1.1)
PCR (Section 1.1.1)
IDT reagent
Insertions >100 bases
Primer Extension (Section 1.1.2) Primer Extension (Section 1.1.2)or Inverse PCR (Section 1.1.3)
IDT reagent
Ultramer™ Oligonucleotides
Deletions <50 bases
Inverse PCR (Section 1.1.3) or
Primer Extension (Section 1.1.2)
Primer Extension (1.1.2)
IDT reagent
Ultramer™ Oligonucleotides
Deletions >50 bases
Inverse PCR (Section 1.1.3)
Primer Extension (Section 1.1.2)
IDT reagent
25 nmole desalted oligos
25 nmole desalted oligos
Table 1. Site-directed Mutagenesis Methods and Outcomes
1.1 Site-directed Mutagenesis
Site-directed mutagenesis creates a mutation at a defined site and requires a known template sequence. This method of altering the sequence allows researchers to investi-gate the impact of sequence changes, such as single nucleotide polymorphisms (SNPs), or to insert or delete a sequence element, such as a ligand binding site or restriction site. Alternatively, site-directed mutagenesis can be used to screen a variety of mutants to determine the optimal sequence for the question at hand.
Site-directed mutagenesis is typically performed using PCR. The identification and con-struction of new commercial polymerases and advances in oligonucleotide synthesis have dramatically increased its efficiency. Primers designed with mutations can intro-duce small sequence changes, and primer extension or inverse PCR can be used to achieve longer mutant regions. In addition, PCR has provided increased precision along with a decrease in cost and time spent on mutagenesis experiments [3]. While the rest of this section will discuss the various PCR methods for site-directed mutagenesis, Section 2 will show how some of these mutation strategies can be more easily accomplished using long oligonucleotides called Ultramer Oligonucleotides.
1.1.1 PCR for Substitutions, Additions, and Deletions
Changes to sequence can be made using PCR by simply including the desired change in one of the PCR primers [4]. The changes can be base substitutions (Figure 1A), additions, or deletions (Figure 1B). The primers are designed to include the desired change. As the primers are extended in the PCR, the resulting amplification product incorporates the mutation, replacing the original sequence. The method is typically very efficient, but can be adversely affected by impure oligonucleotide primers. (See Oligonucleotide Quality Requirements for Mutagenesis Protocols, page 22, for more information on the impor-tance of oligonucleotide purity in mutagenesis).
In addition to internal changes, terminal additions can also be accomplished by PCR. This process is also known as "mispriming", and was first described by Mullis and Faloona [5]. The process uses a primer with additional 5' sequence not complementary to the target. The extension is added to the new product sequence during PCR. In this way, additions can be added to either or both terminal ends of a sequence (Figure 1C). The terminal additions must be made on the 5' end of the primer; any extensions on the 3' end of the primer would result in PCR failure and, therefore, cannot be done using ter-minal addition by PCR.
While PCR for substitutions, additions, and deletions is a simple way to introduce a muta-tion, it is limited by the fact that the mutation can only be introduced in the sequence covered by the primers rather than the sequence that lies between the primers [3].
Mutagenesis Application Guide
Example: Substituting bases in a sequence
A primer that contains the complementary bases for the desired sequence is used in a PCR. All other bases in both primers are complementary to the existing sequence. The PCR will amplify the new sequence and the final product will have the desired sequence with the base changes.
Figure 1A. PCR for Base Substitutions. Primers containing the base changes of interest as a non-comple-
mentary break in the primer sequence (indicated by the blue bubble in primer A) are used in a PCR reac-
tion. As the primers are extended, the resulting amplification product incorporates the mutation, replac-
ing the original sequence (shown as a blue bar in the PCR product).
Figure 1B. PCR for Deletions. Primer A contains complementary sequence to the regions flanking the area to
be deleted. During PCR, primer binding will cause a region of the template to loop out, and amplify only the
complementary region. The final product is shorter because it is missing the deleted sequence.
Figure 1C. PCR for Terminal Additions. An Ultramer™ primer containing an addition to the sequence on the
5' end (the 6X His tag, primer B) is used along with the complementary primer A to amplify a new product
containing the terminal addition.
Mutagenesis Application Guide
1.1.2 Primer Extension
Site-directed mutagenesis by primer extension was first described by Ho et al. and involves incorporating mutagenic primers in independent, nested PCRs to ultimately combine them in the final product [6]. The reaction uses flanking primers (primers A and D) on either end of the target sequence, plus two internal primers (primers B and C) that contain the mismatched or inserted bases and hybridize to the region where the muta-tion will occur. The first round of PCR creates the AB and CD fragments. The two PCR products are mixed together for a second round of PCR. Because primers B and C have complementary ends, the two fragments will hybridize in the second PCR with primers A and D. The final product AD will contain the mutated sequence (Figure 2A).
Figure 2A. Primer Extension for an Insertion.
Primers B and C contain the complementary
sequence that will be inserted (indicated by
the blue line). The first round of PCR uses two
reactions with primer pairs A/B (1) and C/D (2). The
two resulting PCR products are mixed together
with primer pair A/D for a second round of PCR.
The overlapping regions of the two, first-round
PCR products allow the strands to hybridize and
the second round of PCR creates the final, full-
length product with the desired insertion.
Site-directed mutagenesis using primer extension for base deletions or changes is a similar process to that just described. To create a deletion, the B and C primers are posi-tioned on either side of the region to be deleted so that it does not become part of the AB and CD fragments. By design, the B and C primers include complementary sequence so that the AB fragment will hybridize to the CD fragment for the final extension. Be-cause the wild type sequence is not amplified, the final AD product will not contain the deleted fragment (Figure 2B) [6].
Primer extension for additions requires that any additional bases must reside within one of the primers. Traditionally, primer length was limited to 30–50 bases due to synthe-sis yield constraints. However, with the advent of improved synthesis technology, use of especially long primers, such as the IDT Ultramer Oligonucleotides, now makes longer additions possible.
Figure 2B. Primer Extension for Deletions. Prim-
ers B and C are located on either side of the se-
quence to be deleted and contain sequence from
both sides of the deletion (indicated by black or
gray additions that match the black or gray origi-
nal sequence). This sequence will allow them to
overlap with the other fragment after the first
round of PCR. The first round of PCR uses primer
pairs A/B and C/D. The two resulting PCR products
are mixed together with the primer pair A/D for
a second round of PCR. The overlapping regions
of these two, first-round PCR products allow the
strands to hybridize and the second round of PCR
creates the final, full-length product with the de-
sired area deleted.
Alternatively, Lee t al. recently described a new method, overlap extension PCR, that in-volves an additional set of PCR primers to bring in a longer insert [7]. Primer sets A/B and E/F are used to amplify the original sequence while primer set C/D is used to amplify the insert cassette. Primers B, C, D, and E have additional overlapping sequence that, when hybridized, align the three sections in the correct order. Thus, during a second round of PCR using the primer set A/F, the three separate pieces are incorporated together (Figure 2C).
Figure 2C. Primer Extension for Longer Additions.
Primer sets A/B and E/F are used to amplify the
original sequence into two fragments while prim-
er set C/D is used to amplify an insert cassette.
Primers E and B contain additional complemen-
tary sequence to the insert cassette (indicated by
the blue line) and Primers C and D contain addi-
tional complementary sequence to the original
sequence (indicated by the black or gray additions
that match the black or gray original sequence).
The three resulting PCR products are mixed to-
gether with primer pair A/F for a second round
of PCR. The overlapping regions of the three, first-
round PCR products allow the strands to hybrid-
ize and the second round of PCR creates the final,
full-length product with the original sequence
and the inserted cassette.
Mutagenesis Application Guide
One of the most widely adopted methods for introducing changes by primer extension was developed by Stratagene and is marketed as the QuikChange® Site-Directed Muta-genesis Kit. This mutagenesis protocol requires two complementary oligonucleotides, a high-fidelity polymerase, and the restriction enzyme, DpnI. The approach is to hybridize complementary oligonucleotides that contain altered sequence at their center to de-natured double-stranded plasmid DNA. A high-fidelity polymerase is used to generate a copy of each strand of the plasmid DNA by priming from the mutagenic primers. This polymerase does not displace the newly synthesized strands and so the extension stops when the primers copy the entire plasmid and return to the 5' end of the primer. The extension mix is treated with the DNA endonuclease restriction enzyme, DpnI, which requires that the N7 position of adenine be methylated as part of its GMATC recognition sequence. The methylated adenine is only present on the parental plasmid (due to the action of the bacterial DNA methyltransferase, Dam). Thus, DpnI selectively cleaves the parental plasmid DNA, leaving only the mutagenic strands. Once transformed into high efficiency competent bacteria, the annealed mutagenic strand nicks are sealed and the plasmid, now carrying the mutation, is replicated.
1.1.3 Inverse PCR
While traditional PCR amplifies a region of known sequence, inverse PCR uses primers oriented in the reverse direction to amplify a region of unknown sequence [8]. Muta-genic primers can be used to change cloned sequences using a technique adapted from the inverse PCR method [9]. In this method, the entire circular plasmid is amplified and a sequence is deleted (Figure 3A), changed (Figure 3B), or inserted (Figure 3C). The primers are positioned ‘back-to-back', facing outward, on the two opposite DNA strands. One or both of the primers contain the mismatches to create the desired mutations, and both may also carry phosphorylated 5' ends or a restriction site for subsequent recirculariza-tion. The DNA polymerase used in the PCR must be high fidelity and leave blunt ends for subsequent ligation. After PCR, the plasmid DNA is purified, the phosphorylated ends are ligated, and the recircularized plasmid is transformed. [9].
An updated version of this method with a protocol for its use was recently published by Erster and Liscovitch [10]. The authors suggest this method could be used for many types of insertions into target proteins including: ligand-binding domains, functional domains, cleavage sites, tags, and regulatory elements.
Figure 3A. Inverse PCR for a Deletion. This method uses primers that hybridize
to regions on either side of the area to be deleted. In this case, the primers
contain 5' phosphorylated ends to allow the two ends to be ligated together
following amplification. PCR with a high fidelity DNA polymerase that leaves
blunt ends creates a linearized fragment that is missing the deleted region.
This fragment is then recircularized by intramolecular ligation and the result-
ing plasmid is transformed.
Mutagenesis Application Guide
Figure 3B. Inverse PCR for a Substitution. One of the two primers contains the
mutation of interest (indicated by the blue bubble). In this case, both primers
contain 5' phosphorylated ends to allow the two ends to be ligated together
following amplification. PCR is used to amplify the entire circular plasmid to
create a linear template that contains the substituted sequence. This fragment
is then recircularized by intramolecular ligation and the resulting plasmid is
Figure 3C. Inverse PCR for an Insertion. The primers are lined up back-to-back
on either side of the area where the new sequence will be inserted (indicat-
ed by the black, dotted line). One of the primers contains the additional se-
quence that will be inserted (indicated by the blue line). Both primers contain
5' phosphorylated ends to facilitate ligation following amplification. PCR cre-
ates a linearized fragment containing the new sequence. The plasmid is then
recircularized by intramolecular ligation and transformed.
Mutagenesis Application Guide
1.1.4 Cassette MutagenesisCassette mutagenesis replaces a section of DNA sequence with a DNA fragment con-taining the mutated sequence [2]. The insert and target fragments can be generated ei-ther by restriction digestion, PCR amplification, or commercial synthesis of products like IDT Ultramer Oligonucleotides or IDT Genes. The restriction digestion method is depen-dent on the presence of restriction sites on each side of the area that will be replaced. These restriction sites may be present in the original plasmid or may be inserted into its sequence [11]. In this method, the original plasmid is cleaved on either side of the region of interest with two restriction enzymes (Figure 4). The new cassette is then ligated into the linearized plasmid. The PCR amplification method does not require specific restric-tion sites but does require synthetic oligonucleotides.
Cassette mutagenesis has many advantages. It is a very efficient technique and provides an easy way to screen mutants by simply sequencing the resulting plasmids. It provides flexibility to perform many different mutagenesis events on the same vector once set up with flanking restriction sites. Multiple cassettes can also be inserted throughout the vector [12]. Disadvantages to this approach include the need to identify or insert appro-priate restriction sites for exchanging cassettes. Unless the mutated cassette is obtained commercially, the mutations must be generated using oligonucleotides, cloned, and se-quenced before insertion back into the plasmid. Finally, cassette mutagenesis can lead to occasional double mutants or deletion mutants resulting from oligonucleotide impurities, underscoring the importance of using high quality primers. (See Oligonucleotide Quality Requirements for Mutagenesis Protocols, page 22, for more information on the impor-tance of oligonucleotide purity in mutagenesis).
1.2 Random Mutagenesis (in vitro saturation)Random mutagenesis creates mutations at undefined sites and does not require knowl-edge of sequence or function. This approach is a powerful means to identify protein variants with desired properties as well as to understand the interaction of genes in biological pathways. Using a screening assay designed to detect a specific phenotype, protein sequence libraries can be put through multiple rounds of mutation and selec-tion in a process known as directed protein evolution. Through this process, investiga-tors can reveal the function of proteins by mapping enzymatic active sites, studying their role in cellular events, or examining structure-function relationships [13]. If multiple desired mutations are identified in the same gene, investigators can use site-directed mutagenesis to combine these mutations to test their interactions with other genes in biological pathways [14].
Figure 4. Cassette Mutagenesis. The original plasmid is cleaved with restric-
tion enzymes A and E on either side of the cassette to be removed (indicated
in orange). The restriction digest creates a linearized plasmid fragment and a
cassette. The new cassette (indicated in blue) containing the desired changes
is then ligated into the linearized plasmid.
Mutagenesis Application Guide
Random mutations can be introduced in a number of ways including by chemical mu-tagenesis, error-prone PCR (enzymatic mutagenesis), PCR with degenerate primers, UV irradiation, mutator strains, nucleotide analogs, or DNA recombination [15]. Three of these—error-prone PCR, PCR with degenerate primers, and chemical mutagenesis—are discussed here in more detail.
1.2.1 Error-prone PCR
Error-prone PCR is the standard method that researchers use to create libraries of mu-tations within single genes. The procedure is simple and is used in the most common type of mutagenesis experiments: those that examine a small number of mutations to identify a desired phenotype. DNA polymerases are not 100% efficient and have varying degrees of fidelity. Of those tested, Taq DNA polymerase has the lowest fidelity, with an error rate of 0.001–0.02% per nucleotide per pass of the polymerase [16, 17]. For most re-action conditions, this error rate is not sufficient to cause mutagenesis. However, altering the reaction conditions, the polymerase, or the divalent cation used by the polymerase can increase the error rate and generate mutations [18]. Cadwell and Joyce reported increasing the mutation rate to 0.66% ± 0.13% per position per PCR [18]. The advan-tages to this method include the ability to repeat mutagenesis through many rounds of selection as well as to create mutant libraries from randomized cloned genes in order to screen for specific phenotypes. A caveat is that Taq polymerase has a bias toward inducing mutations in A and T bases [19]. Further, excessively altered conditions lead to poor amplification and undesired amplicons; thus, a balance must be achieved between conditions ideal for mutagenesis and those that lead to PCR artifacts [18].
The area that undergoes mutagenesis in error-prone PCR is determined by the position of the primers. The actual reaction is performed under conditions that reduce the fidel-ity, or increase the error rate, of the polymerase. Thus, the number of reaction cycles determines the degree of mutagenesis [20].
A new technique, called MutaGen™, improves on this method by adding a selective PCR amplification of the replicated mutated sequences following the initial round of mutagenic replication [21]. The combination of the two steps allows a wider range of variants to be obtained and combines all types of mutations, including codon deletions. MutaGen™ has been reported as an efficient method for generating libraries with hu-man fragment antibodies, or those displaying different mutation rates and complemen-tary mutational spectra. The diversity of mutations can be increased further by creating libraries using different DNA polymerases and then pooling them into a single library.
1.2.2 PCR with Degenerate Primers
PCR with degenerate primers can also be used for semi-random mutagenesis. In this approach, primers are synthesized with a mixture of wild type and non-wild type nu-cleotides which results in predictable rates of misincorporation per nucleotide [22]. The primers can also contain both regions of wild type sequence and degenerate sequence. Degenerate regions are synthesized with a mixture of the nucleotides consisting primar-ily of the wild type base with a small percentage of the other three bases. Oligonucle-otides with mixed base composition can be ordered from commercial vendors like IDT.
The degenerate PCR method is the most cost-effective method for performing satura-tion mutagenesis—generation of all possible mutations within a gene region—due to the shorter oligonucleotides used [22]. In addition, degenerate PCR also biases against mutants with multiple mutations when there is a single degenerate base position. This makes it a good tool for identifying single base changes that alter function. Because the percentage of base substitutions at each position in the primer can be controlled, this method allows for more precise control over the location and rate of mutagenesis than other random mutation methods. The disadvantages to this method are that it can bias mutations toward sequences with a higher binding affinity for the degenerate primers and changes are limited to the primer binding locations. In addition, it is often hard or impossible to select for changes that will result in a subset of amino acids.
1.2.3 Chemical Mutagenesis
Chemical mutagenesis can induce changes in living cells across the entire genome. These changes avoid the bias that PCR-based mutagenesis methods have toward AT to GC transversions as a result of DNA polymerase bias. In addition, this method has the advantage of selecting for non-lethal mutations because the cells must replicate for the changes to be observed. Lai et al. first described use of ethyl methane sulfonate (EMS) for in vitro mutagenesis in the coding region of a gene [23]. EMS is an alkylating agent that results in G-T mismatches through introduction of AT to GC and GC to AT transition mu-tations. The degree of mutagenesis achieved can be altered by changing the reactions conditions, such as concentration of EMS, incubation time and temperature, reaction pH, or the length or amount of the targeted gene.
Mutagenesis Application Guide
IDT Product Focus: Reagents for Mutagenesis
PrimersIDT offers custom DNA synthesis on scales from 25 nmole to 10 μmole. Every oligo-nucleotide primer is deprotected and desalted to remove small molecule impuri-ties. Oligonucleotides are quantified twice by UV spectrophotometry to provide an accurate measure of yield and are quality control checked by mass spectrometry.
Ultramer™ OligonucleotidesUltramer Oligonucleotides are 25–200 bases long and are synthesized using IDT proprietary, high-fidelity synthesis systems and chemistries. They are the longest, highest-quality oligonucleotides commercially available and are ideal for demand-ing applications like cloning, ddRNAi, and gene construction. Researchers can save a great deal of time and trouble in these applications through direct synthesis of the entire target fragment. Ultramer Oligonucleotides are available on several scales, and can come with attached modifications such as 5' phosphate, biotin, and amino modifiers C6 and C12. Internal degenerate bases, as well as deoxyuracil and deoxyInosine modifications are also available.
Phosphate ModificationsPhosphate modifications may be added to any primer or Ultramer Oligonucle-otide. 5' phosphorylation is necessary if the product will be used as a substrate for DNA ligase, as when two pieces will be ligated together to create a combined, lon-ger product. 3' phosphorylation will inhibit degradation by some 3'-exonucleases and can be used to block extension by many DNA polymerases.
Genes IDT provides a confidential custom gene synthesis service. By ordering genes from IDT, researchers not only save money spent on reagents necessary for construc-tion, cloning, and sequencing, but can also save time by outsourcing the manu-facturing of hard-to-clone gene sequences which often result in repeated failures. At IDT, all genes are constructed using Ultramer Oligonucleotides and the highest fidelity next generation synthesis technology available. Genes arrive in a plasmid cloning vector and are ready for use in a variety of applications.
For more information and to order these products, please visit IDT's website at www.idtdna.com.
IDT Product Focus: Degenerate Primers
Machine Mixed Bases Machine mixed bases contain an equal ratio of each base and are used to create random primers. To order, enter the IUB symbols (e.g., R for a mix of A and G) into the sequence on the IDT DNA ordering page (www.idtdna.com/order). For a com-plete list of the IUB symbols, see the Mixed Bases tab on the DNA ordering page.
Custom Mixed Bases Custom mixes of bases allow customers to specify the ratio of each base. Both hand mix and machine mix options are available. To order click on the Mixed Bases tab on the IDT DNA ordering page (www.idtdna.com/order).
TrimersInserting serial N bases gives rise to all 64 possible codons. This does not pro-duce an equal representation of the 20 amino acids (AAs), but rather biases toward those AAs that have more codons encoding them. Additionally, serial N bases will also insert unwanted stop codons. To avoid this, a set of trimer phosphoramidites have been developed which comprise a single codon for each of the 20 AAs. These are available as a 20 Trimer Mix for creating better N-domains in oligonucleotides intended to encode proteins. It is also possible to obtain custom mixes with more limited amino acid content. To add Trimers to your oligonucleotide order, click on the modification tab on the IDT DNA ordering page (www.idtdna.com/order).
Universal basesTo add these modifications to your oligonucleotide order, click on the modification tab on the IDT DNA ordering page (www.idtdna.com/order).
DeoxyInosine is a naturally occurring base that, while not truly universal, is less destabilizing than mismatches involving the 4 standard bases. Some base pairing bias does exist with dI:dC > dI:dA > dI:dG > dI:dT. When present in a DNA template, deoxyInosine preferentially directs incorporation of dC by DNA polymerase into the growing nascent strand.
5-Nitroindole is currently the best universal base available. It does not favor any particular base pairing and is not as destabilizing to the duplex as mis-matches between the standard bases. 5-Nitroindole directs random incorpo-ration of any specific base when used as a template for DNA polymerase, and partially blocks enzyme processivity.
Mutagenesis Application Guide
Oligonucleotide Quality Requirements forMutagenesis Protocols
For mutagenesis applications, the quality of the oligonucleotide primers is a criti-cal consideration. Impure oligonucleotides can adversely affect the reaction effi-ciency and can introduce additional undesired mutations. IDT monitors every cus-tom synthesis reaction on every synthesis platform and maintains a base-coupling efficiency that is higher than the industry standard. IDT has also pioneered the use of high-throughput quality control (QC) methods and is the only oligonucleotide manufacturer that offers 100% QC and purity guarantees. QC documents are even made available to customers. In addition, IDT evaluates product quality compared to competitor products; IDT oligonucleotides consistently rank as the most pure. This exceptional oligonucleotide quality reduces downstream processing costs, such as assembly and sequencing, and lowers the overall cost of generating se-quences carrying mutations.
In addition to purity, IDT tests its oligonucleotides against those from competitors in functional studies. A recent performance test examined primers used for site-directed mutagenesis (SDM). Four pairs of SDM primers were ordered from four dif-ferent companies (including IDT). These sets were:
Set 1 = Single base change C to G (40mers)
Set 2 = Random 20 bp mutagenesis (60mers)
Set 3 = Addition of a 20 bp section of the repetitive element GGT (60mers)
Set 4 = Deletion of a 20 bp section (60mers)
These oligonucleotides were used in parallel SDM experiments, and resulting clones were screened by IDT scientists. The data from the cumulative cloning experiments show that, in every case, use of IDT oligonucleotides led to better mutagenesis results.
Correct Colonies out of 8 Tested
Competitor A
Competitor B
Competitor C
2. Mutagenesis with Ultramer™ OligonucleotidesUltramer primers are oligonucleotides that range in length from 25–200 bases and sim-plify the addition of large changes into a target sequence. These high-fidelity oligonu-cleotides are the longest oligonucleotides commercially available. Coupling efficiencies for their synthesis routinely reach 99.7%, which eliminates the need for purification.
Ultramer primers are especially useful in PCR and site-directed mutagenesis reactions. Incorporating Ultramer Oligonucleotides as the mutagenic primers allows for greater flexibility in the type and size of sequence changes that can be made. The 3' end of the Ultramer Oligonucleotide primes the PCR with the additional sequence placed upstream (5'). Researchers can add long stretches of new sequences to an existing clone by adding up to 180 bases to a 20 base PCR primer, make changes to a large area within a clone in a single reaction, or change or correct multiple sequence locations simultaneously (Figure 5). Ultramer primers also provide a new tool for regions that have traditionally been difficult to target by extending primers into regions with more optimal sequence composition than was previously attainable.
Changing sequence identity at the 5' and 3' ends of a known sequence, or adding ad-ditional flanking sequences, are the most straightforward ways to modify DNA. Typically, this procedure is used for adding terminal restriction sites to aid in cloning, adding pro-tein purification tags such as 6X His, or adding bacterial phage promoters such as T7/T3/SP6 for synthesis of in vitro RNA transcripts. However, site-directed mutagenesis makes it possible to introduce sequence changes anywhere within a cloned sequence. Here we provide design recommendations for using Ultramer primers with these types of muta-genesis procedures as well as example protocols. The first techniques, Terminal Changes by PCR and Terminal Additions by PCR, create changes at the ends of a PCR amplicon. The second approach, Oligonucleotide-directed Internal Mutagenesis, creates changes anywhere within a cloned sequence.
Example: Using Ultramer Oligonucleotides for Site-Directed Mutagenesis
Ultramer Oligonucleotides allow mutations to be made over a large region in a single PCR extension reaction. In the experiment in Figure 5, a 33-base region was targeted for site-directed mutagenesis with Ultramer Oligonucleotides that had degenerate bases added at specific points. The oligonucleotides were desalted, but not purified further. The degenerate bases allowed a set of these long oligonucleotides to create a library of clones containing a variety of mutations in the targeted region. Each of the lines of sequence represent single clones that were created. Although a few clones do have

Mutagenesis Application Guide
mutations outside the 33-base region, almost all only contain mutations in the target-ed area. This experiment exhibits both how Ultramer Oligonucleotides can be used for creating multiple mutations in a broad region and how additional purification of these products is not mandatory.
Figure 5A. 93mer Ultramer™ Primers Used for Mutagenesis (Above left) Aspartic acid residue 7 and glutamic
acid residue 8, contribute to the active site of the Pyrococcus abysii RNase H2 enzyme. Targeted saturation site-
directed mutagenesis was carried out on residues 2–12 using a 93mer Ultramer™ Oligonucleotide. 2 desalted
complementary Ultramer™ Oligonucleotides were synthesized with 30 base non-degenerate 5' and 3' ends. The
internal sequences of the Ultramer™ Oligonucleotides were synthesized with mixed phosphoramidites at a ratio
of 91:3:3:3 so that mutations would be introduced but not at every site. 2 desalted 93mer degenerate Ultramer™
Oligonucleotides were run on a 10% acrylamide, 7 M Urea denaturing gel and visualized with GelStar® Nucleic
Acid Gel Stain (Cambrex Bio Science Rockland). The size markers were PAGE purified Ultramer™ Oligonucleotides
of 80, 100, 125, 150, 175, 200, and 225 nt in length.
Figure 5B. Sequence of RNase H2 Enzyme Active Site Mutations (Above right) The mutagenesis reaction was
completed by extension with KOD DNA polymerase, followed by DpnI digestion of the parent plasmid, and
transformation into competent BL21DE3 cells. 59 of the resultant clones were sequenced. Even though the
Ultramer™ Oligonucleotides were not purified further than standard desalting, 88% of the clones contained
only mutations within the central 11 amino acid targeted region. The majority of these clones had between 3–5
mutations within this region.
2.1 Ultramer Primer Design
Primer design and cycling conditions will vary based on the type of sequence change you intend to make. While some of the basic PCR primer design rules hold true, others may have to be amended as the template sequence places some constraints on the primer sequence.
Accurately calculating the melting temperature (Tm) of the oligonucleotide primers, minimizing primer-primer interactions, starting with the correct amount of template, and carefully screening against potential off-target hybridization are all key factors to a successful PCR amplification. You can easily calculate an accurate Tm and screen the se-quence for potential interactions (e.g., dimer formation) with the free IDT online SciTools® software, OligoAnalyzer® 3.1 (www.idtdna.com/OligoAnalyzer). To calculate the Tm of the oligonucleotide, you need to know the final monovalent salt (K+, Na+, NH +
4 ), divalent salt
(Mg2+), dNTP, and oligonucleotide concentration of the planned PCR. These values must be entered into the input parameters in OligoAnalyzer 3.1 to get an accurate calculation of the Tm. Unfortunately, many vendors do not disclose their PCR buffer composition. If that information is not available, use default conditions of 50 mM [K+]. When the magne-sium salt is included in the buffer, its concentration is typically at 1.5–2 mM, except for buffers used for qPCR where the [Mg2+] starts at 3 mM.
The primer sequence should have little to no secondary structure. You can measure the stability of any secondary structure within the oligonucleotide sequence with UNAfold, another free tool within the online suite of IDT SciTools (www.idtdna.com/SciTools). In the output, confirm that the Tm of the folded sequence is at least 5–10°C less than the annealing temperature and the secondary structure has a ΔG value between 0 and -9 kcal/mol. For additional discussion, see Troubleshooting Section 3.1.1 Good Primer Design.
IDT Product Focus: Ultramer™ Oligonucleotides
Ultramer Oligonucleotides are 25−200 bases long and are synthesized using IDT proprietary, high-fidelity synthesis systems and chemistries. They are the longest, highest quality oligo-nucleotides commercially available and are ideal for demanding applications like cloning, ddRNAi, and gene construction. Researchers can save a great deal of time and trouble in these applications through direct synthesis of the entire target fragment. Ultramer Oligo-nucleotides are available on several scales, and can come with attached modifications such as 5' phosphate, biotin, and amino modifiers C6 and C12. Internal degenerate bases, as well as deoxyuracil and deoxyInosine modifications are also available.
Mutagenesis Application Guide
2.1.1 Terminal Changes by PCR
See Section 2.3.1 for an example protocol for this method.
Designing the oligonucleotide primers for this type of mutagenesis is straightforward. If the goal is to simply change one to a few bases (e.g., to add or remove a restriction site), then alter the primer sequence to reflect the desired sequence, calculate the Tm of the mismatch using OligoAnalyzer 3.1, and use an annealing temperature 1–2°C lower than the calculated Tm for the mutagenic primer. Sequential base changes will have a greater destabilizing effect and will significantly lower the melting temperature.
Example: Changing a base near the 5' end of a sequence to generate a Bam HI (GGATCC) restriction site.
The Tm was calculated using OligoAnalyzer 3.1 with the following conditions: 0.25 μM oligo, 50 mM KCl, 2 mM MgCl2, 0.8 mM dNTP. Cycling conditions used with the hot start KOD DNA Polymerase (0.5 U) (Novagen) were 2 min 95°C; 30 x (20 sec 95°C, 15 sec 56°C, 45 sec 70°C).
Desired mutation underlined red
Mutagenic forward primer
GgATCCAAGCTGTAATGCTCTA
Wild type sequence
GCATCCAAGCTGTAATGCTCTA
If the PCR product will be digested with a restriction enzyme and subcloned directly, add 3–5 T bases at the 5' end of the primer to allow for efficient binding and cleavage of the PCR product by the restriction enzyme. Do not include those additional bases in the T calculation.
2.1.2 Terminal Additions by PCR See Section 2.3.1 for an example protocol for this method.
To design the primers, select the sequence by extending the length base-by-base from the 5' end of each DNA strand until the calculated Tm of the proposed primer sequence matches the desired annealing temperature for the PCR reaction, typically in the range of 58–65°C. The oligonucleotide concentration, monovalent salt concentration, dNTP concentration, and Mg2+ ion concentration are necessary for accurate Tm calculations. Exclude primer designs that form stable heterodimers, homodimers, or hairpins, if pos-sible. While it may not be possible to completely eliminate such undesired interactions, increasing or decreasing primer length should minimize these interactions. Potential interactions with heterodimers, homodimers, and hairpins can be evaluated using Oli-goAnalyzer 3.1 (www.idtdna.com/OligoAnalyzer). Once the forward and reverse primers have been designed, add the new sequence in front of the appropriate primer. The PCR thermocycling profile is based on the initial annealing temperature of the primer with-out the additional sequence added.
Example: Adding HSV-Tag, 6X His tag to the 3' end of the coding sequence for the Pyro-coccus abysii RNase H2 gene.
Target sequence:
The following primers were selected. Tm was calculated using OligoAnalyzer 3.1 with the following conditions: 0.25 μM oligo, 50 mM KCl, 2 mM MgCl2, 0.8 mM dNTP.
Extended reverse primer with HSV-Tag (in green) and 6X His tag (in red)5′-TCAGTGGTGGTGGTGGTGGTGCTCGACATCCTCGGGGTCTTCCGGGGCGAGTTCTGGCTGGCTgttgcgaaaa
Mutagenesis Application Guide
2.1.3 Oligonucleotide-directed Internal Mutagenesis
See Section 2.3.2 for an example protocol for this method.
As discussed in Section 1.1.2, one of the most widely adopted methods for introducing changes anywhere within a plasmid was developed by Stratagene and marketed as the QuikChange® Site-Directed Mutagenesis Kit. Incorporating Ultramer primers into this method is both simple and highly effective.
The Ultramer primer sequence is designed by selecting 25 bases upstream and down-stream (shown underlined, below) of the site to be mutagenized (shown in red, below). The goal is to get the Tm of this flanking sequence to be 60°C or greater. Add in the desired sequence (shown in blue, below) to finalize the Ultramer primer sequence, and create the reverse complement sequence. The Stratagene protocol recommends puri-fied primers but this step is not necessary when high-fidelity Ultramer Oligonucleotides are used—desalted oligonucleotides of this length do not require additional purification as long as more than one clone will be sequenced.
Example: Mutagenic oligo designAspartic acid residue 7 and glutamic acid residue 8, contribute to the active site of the Pyrococcus abysii RNase H2 enzyme. Amino acids Asp7 and Glu8 (highlighted in red) were targeted for muta-genesis to Arg and Lys. 25 bases upstream and downstream of the site to be mutagenized (shown underlined) were selected. The desired sequence (highlighted in blue) was added and the reverse complement sequence was created.
CAACaagctt----plasmid vector sequence
2.2 Controls
Effective controls are an integral component of any mutagenesis experiment. The con-trols listed below will confirm each step in the process toward making a site-specific mu-tation. You may choose to eliminate some of these controls but it is important to weigh removing a control against the time and costs associated with repeating the experiment if you observe incomplete or poor results.
At a minimum, each step of the process should include a positive and a negative control. The positive control should be a plasmid of similar size to the experimental plasmid that can be mutated to give an easily identifiable phenotype. This will allow you to follow each of the steps of the mutagenesis reaction with template and primers that are known to work. An example of this type of control would be a plasmid containing a gene ex-pressing the alpha subunit of the β-galactosidase gene. This gene can easily be mutated into an inactive form by changing the start codon to a stop codon, or by adding one or more stop codons to the 5' end of the gene. Detection of this change is easily observed when transformed into cells compatible with blue-white screening (such as DH5α) and plated on agar plates containing X-Gal.
2.2.1 PCR Controls
Negative control—a PCR with all of the PCR components except template. No amplification product should be detectable. If a product is amplified, one of the reagents is contaminated and must be replaced.
Positive control—a PCR containing a control plasmid and primers that will yield a mutant product. This reaction should be run with the same conditions and reaction components as the experimental reaction. The control plasmid should be similar in size to the test sequence so that it will amplify under the same cycling conditions.
When you run these controls on an agarose gel, you should see a strong, single band in the positive PCR control lane and no band in the negative control lane.
Mutagenesis Application Guide
2.2.2 Ligation Controls (If applicable)
Negative control—a reaction containing the positive control from the PCR product and lacking ligase. Once transformed, this reaction will provide an idea of how much background is present in the experimental reaction.
Positive control—a reaction containing the positive control from the PCR prod-uct and containing ligase.
While not routine, gel confirmation of ligation reactions can be carried out if this step is problematic. See Troubleshooting Section 3.6.2 Gel Confirmation of a Ligation Reaction.
2.2.3 DpnI Digestion Controls
Negative control—10 pg undigested, supercoiled plasmid and no DpnI enzyme.
Positive control—10 pg plasmid digested under the same conditions as the experimental PCR product.
2.2.4 Transformation Controls
The following should be transformed into competent cells in the same manner as the experimental sample:
PCR product positive control—Amplify the original plasmid using primers that do not introduce mutations to verify the experimental conditions will produce the expected product. Observing few or no colonies usually indicates a poor amplification and/or poor DpnI digestion.
Ligation positive and negative control—The positive ligation control should have 5–100 times more colonies than the negative ligation control. If this is not the case, see Troubleshooting Section 3.6.3 Inhibitors of Ligase.
DpnI positive and negative control—The ratio of the number of colonies from the DpnI positive and negative control indicates the efficiency of the digestion. The negative control should have 5–100 times more colonies than the positive control. If this is not the case, see Troubleshooting Section 3.7 DpnI Digestion.
A lack of colonies from any of the controls indicates a problem with the competent cells, the transformation protocol, or the media used to select for the correct transformants. If this is the case, see Troubleshooting Section 3.8 Transformation.
2.3 Example Protocols
In this section, we provide two protocols: one for Terminal Changes or Additions and one for Oligonucleotide-directed Internal Mutagenesis. We also provide reaction setup suggestions for two different types of polymerases: Phusion® (New England Biolabs) and KOD (Novagen). These are general protocols and may need to be altered depending on your specific application.
Protocol for Terminal Changes or Additions – see Section 2.3.1, page 31.
Protocol for Oligonucleotide-directed Internal Mutagenesis – see Section 2.3.2, page 35.
2.3.1 Protocol for Terminal Changes or Additions
For more information on this method and primer design considerations, see Section 2.1.2.
1. PCRSet up a 50 μL mutagenesis reaction using a high fidelity polymerase, such as Phusion (New England Biolabs) or KOD DNA polymerase (Novagen). A typical reaction setup and cycling times are described below.
Phusion® (2 U/μL)
KOD (2.5 U/μL)
5X Phusion HF Buffer
Hot Start DNA
Phusion polymerase
dNTPs (2 mM each)
25 mM MgSO4
Forward Primer
Reverse Primer
Total Volume
Mutagenesis Application Guide
PCR Cycling Parameters for KOD DNA Polymerase
PCR Cycling Parameters for Phusion DNA Polymerase
* Time and ** temperature are dependent on
Cycling times and temperatures are shown us-
which high fidelity DNA polymerase is being used
ing Phusion DNA Polymerase and a 4 kb plasmid
and the size of the plasmid being replicated. Cy-
(vector plus insert).
cling times and temperatures are shown using KOD DNA Polymerase and a 4 kb plasmid (vector plus insert). If you are using a different polymerase, follow the manufacturer's recommendation for cycling times.
2. Confirm a full length productRun 5 μL of the reaction on a 0.75% agarose gel.
Reactions that do not produce a single clean band on a gel may require either gel puri-fication or PCR optimization.
3. LigationCircularize the amplicon with T4 DNA Ligase.
This reaction can be carried out at room temperature (22°C) for 5 min if the Quick Liga-tion™ Kit (New England Biolabs) is used. Alternatively, a standard T4 DNA Ligase can be used; follow the manufacturer's instructions. As Taq and other thermostable DNA ligases do not efficiently ligate blunt ended DNA, we do not recommend their use for this application.
2X Quick T4 DNA Ligase Buffer (NEB)
T4 DNA Ligase (NEB)
4. DpnI DigestionAdd 1 μL of DpnI restriction endonuclease (New England Biolabs) to the reaction mix-ture to remove the original plasmid DNA. Incubate at 37°C for 30 min.
5. TransformationTransform 2 μL of the final product into competent E. coli following the manufacturer's instructions. For DNA fragments smaller than 12 Kb, chemically competent cells such as DH5α can be purchased or prepared in the lab. Larger plasmids require electroporation for efficient uptake by bacteria.
Typical transformations use 25–100 μL of competent cells under the following conditions:
1. Place the competent cells on wet ice until completely thawed (2–5 min).
2. Add 2–5 μL of the DpnI digestion product, not exceeding 10% of the competent
cell volume.
3. Incubate on wet ice for 30 min, stirring gently every 10 min. Do not pipette or vortex
4. Place the cells in a 42°C water bath for 30–45 sec.
5. Immediately return the cells to wet ice for 2 min.
6. Add 10 volumes of SOC (e.g., for 25 μL cells, add 250 μL SOC) and place in a shaking
incubator at 37°C for 1 hr. See below for an SOC media recipe.
7. Plate 100–200 μL of the reaction on 10 cm LB agar plates with the appropriate selec-
tion agent (e.g., 100 μg/mL ampicillin or 50 μg/mL kanamycin).
8. Place in an incubator at 37°C overnight.
SOC media (1 liter total volume):
20 g Bacto Tryptone
5 g Bacto Yeast Extract
Mutagenesis Application Guide
6. ScreeningMethods of screening for desired mutations vary depending on the size and complexity of the mutated area. In general, Sanger sequencing using Applied Biosystems BigDye® Terminators and capillary sequencing is the preferred method. A protocol for a rapid pu-rification of DNA to screen colonies is listed below. Alternatively, a plasmid purification kit such as Qiagen's Plasmid Mini Kit can be used. Screen 5–20 colonies to ensure identi-fication of a correct clone. Note that primers used for sequencing should be positioned at least 40 bases away from the mutation site as the first few bases of sequencing reads are often poor.
1. Transfer a colony from the transformation plate to 500 μL LB broth with the appro-
priate antibiotic.
2. Grow shaking at 37°C overnight.
3. Transfer 180 μL to a PCR tube.
4. Spin the tubes at a minimum of 3000 x g for 5 min.
5. Pour off the supernatant and tap the tube dry on a paper towel.
6. Repeat the spin.
7. Add 100 μL of water and vortex.
8. Heat at 90–100°C for 10 min.
9. Spin at 15,000 x g for 5 min.
10. Remove the top 50 μL of the supernatant and use for sequencing (in general 2–5 μL
of this product is sufficient for sequencing when using a high copy number plasmid).
2.3.2 Protocol for Oligonucleotide-directed Internal Mutagenesis
For more information on this method and primer design considerations, see section 2.1.3.
1. PCRSet up a 50 μL mutagenesis reaction using a high fidelity polymerase such as KOD (No-vagen) or Phusion (New England Biolabs). A typical reaction setup and cycling times are described below.
Note: When using the Oligonucleotide-directed Internal Mutagenesis technique, it is essential to use a non-strand-displacing polymerase such as KOD (Novagen), Phusion (New England Biolabs), or Pfu Turbo® (Stratagene). Avoid strand-displacing polymerases such as Taq, Vent, and Deep Vent.
Phusion® (2 U/μL)
KOD (2.5 U/μL)
5X Phusion HF Buffer
Hot Start DNA
Phusion polymerase
dNTPs (2 mM each)
25 mM MgSO4
Forward Primer
Reverse Primer
Total Volume
Mutagenesis Application Guide
PCR Cycling Parameters for KOD DNA Polymerase
PCR Cycling Parameters for Phusion DNA Polymerase
* Time and ** temperature are dependent on
Cycling times and temperatures are shown us-
which high fidelity DNA polymerase is being used
ing Phusion DNA Polymerase and a 4 kb plasmid
and the size of the plasmid being replicated. Cy-
(vector plus insert).
cling times and temperatures are shown using KOD DNA Polymerase and a 4 kb plasmid (vector plus insert). If you are using a different polymerase, follow the manufacturer's recommendation for cycling times.
2. Confirm a full length productRun 5 μl of the reaction on a 0.75% agarose gel.
Reactions that do not produce a single clean band on a gel may require either gel puri-fication or PCR optimization.
3. DpnI DigestionAfter the cycling is finished, add 1 μL of DpnI restriction endonuclease (New England Biolabs) to the reaction mixture to remove the original plasmid DNA. Incubate at 37°C for 30 min.
4. TransformationTransform 2 μL of the final product into competent E. coli following the manufacturer's instructions. For DNA fragments smaller than 12 Kb, chemically competent cells such as DH5α can be purchased or prepared in the lab. Larger plasmids require electroporation for efficient uptake by bacteria.
Typical transformations use 25–100 μL of competent cells under the following conditions:
1. Place the competent cells on wet ice until completely thawed (2–5 min).
2. Add 2–5 μL of the Dpn digestion product, not exceeding 10% of the competent cell
3. Incubate on wet ice for 30 min, stirring gently every 10 min. Do not pipette or vortex
4. Place the cells in a 42°C water bath for 30–45 sec.
5. Immediately return the cells to wet ice for 2 min.
6. Add 10 volumes of SOC (e.g., for 25 μL cells, add 250 μL SOC) and place in a shaking
incubator at 37°C for 1 hr. See below for an SOC media recipe.
7. Plate 100–200 μL of the reaction on 10 cm LB agar plates with the appropriate selec-
tion agent (often 100 μg/mL ampicillin or 50 μg/mL kanamycin).
8. Place in an incubator at 37°C overnight.
SOC media (1 liter total volume):
20 g Bacto Tryptone
5 g Bacto Yeast Extract
5. ScreeningMethods of screening for desired mutations vary depending on the size and complexity of the mutated area. In general, Sanger sequencing using Applied Biosystems BigDye® Terminators and capillary sequencing is the preferred method. A protocol for a rapid purification of DNA to screen colonies is listed below. Alternatively, a plasmid purifica-tion kit such as Qiagen's Plasmid Mini Kit can be used. Screen 5–20 colonies to ensure identification of a correct clone. Note that primers used for sequencing should be at least 40 bases away from the mutation site as the first few bases of sequencing reads are often poor.
1. Transfer a colony from the transformation plate to 500 μL LB broth with the appro-
priate antibiotic.
2. Grow shaking at 37°C overnight.
3. Transfer 180 μL to a PCR tube.
4. Spin the tubes at a minimum of 3000 x g for 5 min.
5. Pour off the supernatant and tap the tube dry on a paper towel.
6. Repeat the spin.
7. Add 100 μL of water and vortex.
8. Heat at 90–100°C for 10 min.
9. Spin at 15,000 x g for 5 min.
10. Remove the top 50 μL of the supernatant and use for sequencing (in general 2–5 μL
of this product is sufficient for sequencing when using a high copy number plasmid).
Mutagenesis Application Guide
3. TroubleshootingMutagenesis is a multi-step process that varies greatly depending on the particular method you choose, the goal of the project, and the information you have about the target sequence. As a result, troubleshooting may be necessary in order to maximize the desired results. Here we list some of the more common issues that arise with site-directed mutagenesis. See the table below for the commonly observed problems and the potential solutions to consider, and then refer to the indicated section to learn more about the issues and how to correct them. You may need to look further into some of these issues than is covered in the scope of this guide. We recommend two additional resources for more information: Molecular Cloning: A Laboratory Manual [24] and Cur-rent Protocols in Molecular Biology [25].
Observed Issue
e deletions or
utants when scr
oo man olonies
utations in plasmids
Multiple P
Too few sat
Colonies in negativ
Plasmids hav rearr
No or few m
3.1 Primer Design
3.2 Template Concentration and Quality
3.3 Reaction Setup
3.4 Reaction Components
3.5 PCR Reaction Parameters
ential PPot
3.7 DpnI Digestion
3.8 Transformation
3.1 Primer Design3.1.1 Good Primer Design
Poorly designed primers can result in a lack of full-length amplification, amplification of multiple products, or no amplification at all. See Section 2.1 for more information on designing primers. Make sure the primers match the target sequence and include the following primer characteristics:
GC content between 35–65%.
Melting temperature (Tm) between 55–65°C.
Tm difference between primers limited to 2–3°C. The primer with the lower Tm will dictate the annealing temperature used in the reaction.
Little dimerization, particularly at the 3' ends where the primer should have no more than 3–4 bases of homology with itself or other primer sequences. However, note that the primers for the QuikChange® method of site-directed mutagenesis will dimerize due to the nature of the experimental design.
No stable hairpin structures—avoid structures with a Tm in the range of the an-nealing temperature.
Note that due to the constraints site-directed mutagenesis imposes on the location of the primers, meeting all of these parameters is not always possible. To evaluate potential primers, use oligonucleotide analysis software like the free online tool, OligoAnalyzer 3.1, located on the IDT website.
IDT Product Focus: SciTools® Design Tools
IDT offers a number of free design and analysis tools on the website. These include:
OligoAnalyzer—for analyzing oligonucleotide melting temperatures, hairpins, di-mers, mismatches, and off-target hybridization
UNAFold—for analyzing oligonucleotide secondary structure
For more information and to access these free SciTools design tools, visit the IDT website (www.idtdna.com/SciTools).
Mutagenesis Application Guide
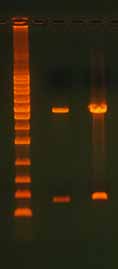
3.2 Template Concentration and Quality3.2.1 Too Much or Too Little TemplateIn any PCR, too much or too little template can have a profound effect on the results. A reaction with too little template typically produces weak or nonexistent bands when the reaction is run on a gel. Too much template can suppress the PCR or result in the production of unwanted end products that may appear as smears on the gel. For most PCR-based applications, high-quality plasmid DNA is used at a concentration of 1–10 ng. Genomic DNA is more commonly used at the 10–100 ng range. In addition to the effects on PCR, loading too much template onto a gel can cause altered band migration, band distortion, and smearing (Figure 6).
Figure 6. Effect of Overloading an Agarose
Gel. Lane 1 has an appropriate amount of DNA
loaded, with 125 ng digested plasmid DNA
(3470 bp). Lane 2 has too much DNA, with 950
ng digested plasmid DNA. In the overloaded
lane, note the altered migration of the lower
molecular weight band, band distortion, and
smear. The ladder is the 1 Kb Ladder DNA Mark-
er (Axygen).
] Altered Migration
3.2.2 Poor Quality TemplateTemplate quality can impact yield and specificity of the PCR product. The presence of impurities such as high concentrations of salts, polysaccharides, dyes, alcohols, and proteins can all affect the PCR by inhibiting the enzyme. The presence of chemi-cals that can sequester the Mg2+ ions, such as EDTA, will also inhibit activity of the polymerase and possibly decrease the efficiency of primer binding. The template DNA can be cleaned up in a variety of ways. Phenol-chloroform extraction followed by ethanol precipitation is a common way to purify DNA. Commercial kits are also available from a variety of vendors.
3.3 Reaction Setup3.3.1 Experimental SetupA failed experiment could be caused by a component inadvertently being left out of the reaction or due to an expired reagent. To minimize pipetting variation and the chance of leaving a reagent out of a reaction, we recommend creating a master mix of reagents common to all reactions in the experiment. Typically the master mix will contain every-thing except the primers and template.
We recommend that you always include a positive control, preferably with an amplicon of similar size to the experimental PCR amplicon, and a non-template negative control. Re-peat a failed experiment at least once to make sure that all components are included. If the positive control does not work, repeat this control while replacing individual components to identify the problematic reagent before trying to optimize the experimental reaction.
Prior to running any PCR, confirm that you have good quality template (see Section 3.2) by visualizing it on an agarose gel. For more information on controls, see Section 2.2.
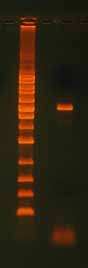
After running any PCR, it is critical to confirm the full length product is present before proceeding to the next step. Run part of the reaction on an agarose gel (Figure 7). Reac-tions that do not produce a single clean band on a gel may require either gel purification or PCR optimization.
Figure 7. QuikChange® Site-Directed Mutagen-
esis Product. After site-directed mutagenesis
reaction, it is important to run the extension
product on an agarose gel to verify that you
have good quality template. Note the 3100 bp
plasmid extension product in this example. If
you do not see a band, the reaction did not
work. The ladder is the 1 Kb Ladder DNA Marker
Mutagenesis Application Guide
3.3.2 Kits
Kits are optimized for use with specific buffers and enzymes. Be sure to use the correct reagents at the suggested concentrations. Note that buffers from different kits are not always interchangeable because the activity of each enzyme is optimal under specific salt and pH conditions.
3.3.3 Controls
Good controls are an essential component to effective troubleshooting. At a mini-mum each PCR should include a positive control that is known to amplify under the same conditions as the unknown and a negative control that lacks template to test for contamination. See Section 2.2 for details on the types of controls to use.
3.4 Reaction Components
A common PCR problem involves using the incorrect concentration of reaction components. Most PCR-based mutagenic techniques have the same basic compo-nents, but they are not always used at the same concentrations. Mg2+ salts are a common example of application-dependent concentration requirements. For most mutagenic reactions, keeping the Mg2+ ion concentration at about 0.7–1 mM is op-timal; however, this amount may vary based on the particular polymerase that is used. Likewise, primer concentrations are typically used in the 100–250 nM range. In contrast, qPCR frequently employs Mg2+ concentrations in the 2–3 mM range, and the primer concentrations can be as high as 900 nM. It is also important to keep in mind that the concentration of free Mg2+ and primers will influence the melting temperature (Tm) of the primers. Increasing concentrations of either component will raise the Tm and can facilitate unwanted side reactions that will consume reagents.
3.4.1 Polymerases
The use of high-fidelity polymerases is preferred over Taq polymerase or other lower fidelity polymerases. High-fidelity polymerases decrease the number of PCR-in-duced errors such as point mutations. When using the Oligonucleotide-directed In-ternal Mutagenesis technique (see Section 2.1.3), it is essential to use a non-strand-displacing polymerase such as KOD (Novagen), Phusion® (New England Biolabs), or Pfu Turbo® (Stratagene), and to avoid strand-displacing polymerases such as Taq, Vent, and Deep Vent.
3.5 PCR Reaction Parameters3.5.1 Cycle Number
Too few PCR cycles may not amplify enough product to be visible on a gel while too many cycles may allow non-specific amplification and increase the chances of poly-merase errors that could result in point mutations or small deletions. For site-directed mutagenesis, it is best to use the minimum number of cycles needed to produce a detectable band when 1/10th of the reaction is analyzed by agarose gel electrophore-sis. Increase or reduce the number of cycles by 3–5 cycles at a time to find the optimal cycle number.
3.5.2 Annealing Temperature
An annealing temperature that is too low allows nonspecific primer binding lead-ing to nonspecific amplification. On a gel, these products appear as a smear or an incorrectly-sized band. An annealing temperature that is too high can result in poor or no primer binding and weak or absent products.
Optimize the annealing temperature by starting with a temperature 2–3°C lower than the calculated annealing temperature of the part of the primer that binds to the template (see primer design in Section 2.1). Change in increments of 2–4°C as necessary.
3.5.3 Extension Time
Different polymerases vary in the rate of extension and the degree to which this rate is affected by secondary structure and sequence complexity. Typical extension rates are between 500–4000 bases per minute. Using extension times that are too short can result in little or no product. Extension times that are too long can result in low product yields due to polymerase denaturation and, in extreme cases, replication of multiple copies of the plasmid.
Follow the polymerase manufacturer's recommendation for the extension time based on the expected size of your amplicon (we recommend starting with the longer end of the extension time if a range is given) and adjust by 15–30 sec incre-ments as needed.
Mutagenesis Application Guide
3.5.4 Denaturation TemperatureTypical denaturation temperatures are 94–95°C for most polymerases. Lower de-naturation temperatures may not completely denature the DNA while higher tem-peratures may reduce the polymerase activity. Follow the manufacturer's guidelines.
3.5.5 Initial Denaturation TimeThe first denaturation step of the PCR should completely denature the plasmid DNA and any proteins carried over from plasmid purification. Thus, this step should be longer than subsequent denaturation steps. A denaturation step that is too short will result in weak or no amplification while an initial denaturation step that is too long may reduce the activity of the polymerase and result in little or no amplifica-tion of the product or controls.
3.5.6 Touchdown PCRNonspecific primer binding may be difficult to prevent if it occurs at a temperature close to the annealing temperature of the desired product. In these cases touch-down PCR may help. In general, touchdown PCR begins cycling with a very high annealing temperature that decreases by 1–2°C for each of the first 5–10 cycles [24]. This allows the preferred binding sites to begin exponential amplification a few steps ahead of closely matching sites.
Create a PCR profile with an annealing temperature 5–10°C above the predicted an-nealing temperature of the primers and decrease it by 1°C per cycle for the first 5–10 cycles. Follow with 20–30 cycles at the lowest annealing temperature.
3.6 Ligation
3.6.1 Quantification of ProductUsing the correct concentration of PCR product is critical for ligation reactions. If the ratio of insert to vector is suboptimal, most of the resultant colonies will be either vector with no insert or other deleted forms of the vector. A concentration that is too low will not produce sufficient product to provide enough colonies for screening. A concentra-tion that is too high (above 10 μg/mL) may favor intermolecular ligation of multiple plasmids, resulting in large products when run on a gel and poor transformation yields. If you observe this, decrease the concentration of the PCR product added to the ligation reaction by 2–10X.
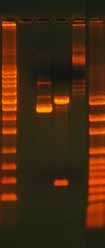
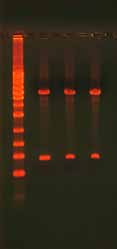
3.6.2 Gel Confirmation of a Ligation ReactionThe success of a ligation reaction can be assessed by running the product on an aga-rose gel (Figure 8). In general, supercoiled products migrate through the gel faster than linear products. Run half of the ligation reaction in a lane next to the linear PCR fragment to determine if a mobility shift has occurred. Note that ligation reactions rarely reach completion so expect the presence of some linear DNA. The high salt content of ligase buffers can also affect the mobility of the product (Figure 9). Purify the product with a NAP5 or similar column if the product shows bands with smears.
Figure 8. Gel Analysis of a Ligation Reaction.
The gel shows 500 ng undigested, supercoiled
plasmid DNA (lane 1), 500 ng digested plasmid
DNA with insert (lane 2), and 500 ng re-ligated,
circular plasmid DNA (lane 3). The ladder on
the left is the 1 Kb ladder DNA Marker (Axygen)
and the ladder on the right is the 100 bp Lad-
der DNA Marker (Axygen). The pGEM plasmid
is 3015 bp and the insert is 400 bp. Note the
differences in how the DNA migrates and the
bands that appear depending on whether the
DNA is undigested (supercoiled), digested, or
re-ligated (circular but not supercoiled).
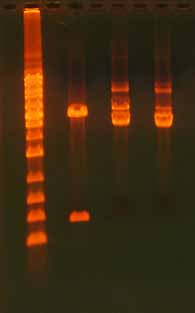
Figure 9. Effect of Salt on DNA Migration Dur-
ing Gel Electrophoresis. Salt is a common com-
ponent of reaction buffers but too much salt
in the sample will effect DNA gel migration.
This gel shows 500 ng digested plasmid DNA
+ 0 mM NaCl (lane 1), 500 ng digested plasmid
DNA + 250 mM NaCl (lane 2), and 500 ng di-
gested plasmid DNA + 500 mM NaCl (lane 3).
The salt was added after the digestion was
complete. Note the "focusing" or "narrowing"
effect and the mobility shift in the presence of
salt. The effect is greater with a higher salt con-
centration. The ladder is the 1 Kb ladder DNA
Marker (Axygen).
Mutagenesis Application Guide
3.6.3 Inhibitors of Ligase
Ligase activity can be reduced or inhibited by several factors including the following:
High levels of salts (Figure 10)—Prior to ligation, desalt the DNA with a clean up kit that has a size exclusion column.
Degraded ATP in the reaction buffer—Aliquot small volumes of the ligase buffer to avoid repeated freeze-thaw cycles.
The use of deoxyribose ATP instead of ribose ATP—Nucleotides for PCR are not an energy source for ligase.
An incubation time that is too short—Blunt-ended ligations often require 2 hours at room temperature or overnight at 16°C for maximum efficiency.
A high degree of secondary structure near the ligation point—this can cause deletions and/or rearrangements in the vicinity of the ligation point as well as poor ligation efficiency.
Poor storage conditions (such as storing in a frost-free freezer) or multiple freeze-thaw cycles.
Figure 10. Inhibitory Effect of Salt
on Restriction Endonuclease Diges-
tion. The gel shows 500 ng digested
plasmid DNA + 0 mM NaCl (lane 1),
500 ng digested plasmid DNA + 250
mM NaCl (lane 2), and 500 ng di-
gested plasmid DNA + 500 mM NaCl
3 Kb vector
(lane 3). The salt was added during
the restriction digest setup and the
restriction enzymes used were SphI
and SacI. Note that the addition of
salt dramatically affects the reaction.
In lanes 2 and 3, the insert is not vis-
ible, the amount of linear template
is reduced, and both supercoiled
400 bp insert
(bottom band) and nicked circular
(top band) products are present. The
ladder is the 1 Kb ladder DNA Marker
3.7 DpnI Digestion3.7.1 Controls
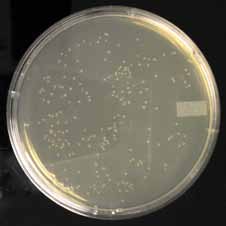
The DpnI digestion can be confirmed by transforming equal amounts of digested and undigested plasmid. An efficient digestion reaction should decrease the number of col-onies by 1–2 orders of magnitude (Figure 11). This control should have the same salts and buffers as the experimental reaction. As with many enzymes, DpnI will become inac-tive when stored at warm temperatures. Store the enzyme in a -20°C freezer.
49 colonies
189 colonies
Figure 11. Effect of DpnI Treatment. The extension product from a site-directed mutagenesis reaction was ei-
ther transformed directly or treated with DpnI and then transformed. All transformations were into chemically
competent bacteria. Colonies shown in plate 1 were derived from reactions treated with DpnI while colonies in
plate 2 were from reactions not treated with DpnI. Note the significantly fewer number of colonies in the DpnI-
treated sample. The extra colonies in plate 2 contain the original plasmid DNA without the mutation of interest.
3.7.2 Methylated DNA
Template DNA requires methylation by the bacterial Dam methylase for subsequent digestion by DpnI. DNA isolated from non-bacterial sources or bacteria lacking Dam methylase, such as the JM110 strain, will not be digested by DpnI and will result in a high background of wild type colonies with few or no mutants.
Mutagenesis Application Guide
3.8 Transformation3.8.1 Handling Competent Cells
Frozen competent cells are very fragile and are sensitive to temperature changes. Once thawed, these cells should not be refrozen as a large loss in competency is likely to occur. After thawing, keep cells on wet ice and use them immediately. Rough handling, includ-ing rapid pipetting and vortexing, will also result in a loss in competency.
3.8.2 Heat Shock Considerations
Many commercially-purchased, chemically-competent bacteria must be subjected to a 30 sec heat shock at 42°C followed by rapid cooling on ice. However, protocols vary regarding heat shock transformation for different cell lines. Choose a protocol that has been successful for the bacterial strain you are using and follow it precisely. Small chang-es; such as the type of tube used, the length of heat shock, or warming the cells even briefly; can have large effects on transformation efficiency.
3.8.3 Electroporation Considerations
Salts, even in small amounts, greatly increase the conductivity of liquids. In electropora-tion this can lead to superheating of the transformation or even arcing of the electro-poration vessel resulting in cell death. Ligation reactions use high salt concentrations so it is important to desalt the DNA prior to electroporation.
3.8.4 Antibiotic Selection
Antibiotic selection is required to eliminate cells that lack plasmids. Useful ranges of antibiotics vary—in general 100 μg/mL ampicillin and 50 μg/mL kanamycin work well to select for high copy plasmids in most E. coli strains.
An antibiotic concentration that is too high can cause death of antibiotic-resistant cells and result in no growth on plates.
An antibiotic concentration that is too low results in growth of non-antibiotic-resistant cells. Often this is seen as a lawn or near lawn of cells when transformation reactions
are plated. Antibiotics degrade over time and are sensitive to heat. Low concentrations of some antibiotics, such as ampicillin, can lead to the growth of satellite colonies. This is because low concentrations of ampicillin are bacteriostatic rather than bactericidal. Beta lactamase, the protein that confers ampicillin resistance, is secreted from cells that express it. As a result, bacterial colonies that lack ampicillin resistance, and are not killed, can grow after the formation of resistant colonies. These smaller satellite colonies lack plasmids and often outnumber the plasmid-containing colonies.
Observation of differently colored cells, filamentous growth, or inhibition of E. coli growth indicates contamination. Replace the media and practice sanitary techniques to avoid this.
3.8.6 E. coli Strains
Many common strains such as DH5α and its derivatives work well for most applications. Plasmid sequences that are large (greater than 8 kb) have high degrees of secondary structure, high GC content, and/or homology to the host genome. The latter factor may allow them to recombine within the genome resulting in deletions and rearrangements, often within one area of the plasmid. The use of E. coli strains designed to have lowered recombination or designed for use with large plasmids can sometimes decrease this recombination. Such strains include the Stbl3 line from Life Technologies and the XL10 gold line from Stratagene.
3.8.7 Toxic Sequences
Some proteins can be toxic to the bacteria when they are expressed from a plasmid. Ex-amples include the sucrase gene and restriction endonucleases. When using mutagen-esis to add new sequences, it is possible to create a toxic protein. A variety of methods can be employed to reduce the toxicity [26].
Mutagenesis Application Guide
4. References
1. Shortle D, DiMaio D, et al. (1981) Directed mutagenesis. Annu Rev Genet, 15: 265–294.
2. Zoller MJ. (1991) New molecular biology methods for protein engineering. Curr
Opin Biotechnol, 2(4): 526–531.
3. Reikofski J, and Tao BY. (1992) Polymerase chain reaction (PCR) techniques for site-
directed mutagenesis. Biotechnol Adv, 10(4): 535–547.
4. Kadowaki H, Kadowaki T, et al. (1989) Use of polymerase chain reaction catalyzed by
Taq DNA polymerase for site-specific mutagenesis. Gene, 76(1): 161–166.
5. Mullis KB and Faloona FA. (1987) Specific synthesis of DNA in vitro via a polymerase-
catalyzed chain reaction. Methods Enzymol, 155: 335–350.
6. Ho SN, Hunt HD, et al. (1989) Site-directed mutagenesis by overlap extension using
the polymerase chain reaction. Gene, 77(1): 51–59.
7. Lee J, Shin MK, et al. (2010) Insertion and deletion mutagenesis by overlap extension
PCR. In: Braman J (editor) Methods Mol Biol New York: Humana Press. 634: 137–146.
8. Ochman H, Gerber AS, et al. (1988) Genetic applications of an inverse polymerase
chain reaction. Genetics, 120(3): 621–623.
9. Hemsley A, Arnheim N, et al. (1989) A simple method for site-directed mutagenesis
using the polymerase chain reaction. Nucleic Acids Res, 17(16): 6545–6551.
10. Erster O and Liscovitch M. (2010) A modified inverse PCR procedure for insertion,
deletion, or replacement of a DNA fragment in a target sequence and its application in the ligand interaction scan method for generation of ligand-regulated proteins. In: Braman J (editor) Methods Mol Biol New York: Humana Press. 634: 157–174.
11. Smith M. (1985) In vitro mutagenesis. Annu Rev Genet, 19: 423–462.
12. Wells JA, Vasser M, et al. (1985) Cassette mutagenesis: an efficient method for gen-
eration of multiple mutations at defined sites. Gene, 34(2–3): 315–323.
13. Georgescu R, Bandara G, et al. (2003) Saturation mutagenesis. Methods Mol Biol, 231:
14. You L and Arnold FH. (1996) Directed evolution of subtilisin E in Bacillus subtilis to
enhance total activity in aqueous dimethylformamide. Protein Eng, 9(1): 77–83.
15. Kuchner O and Arnold FH. (1997) Directed evolution of enzyme catalysts. Trends
Biotechnol, 15(12): 523–530.
16. Eckert KA and Kunkel TA. (1990) High fidelity DNA synthesis by the Thermus
aquaticus DNA polymerase. Nucleic Acids Res, 18(13): 3739–3744.
17. Eckert KA and Kunkel TA. (1991) DNA polymerase fidelity and the polymerase
chain reaction. PCR Methods Appl, 1(1): 17–24.
18. Cadwell RC and Joyce GF. (1992) Randomization of genes by PCR mutagenesis.
PCR Methods Appl, 2(1): 28–33.
19. Moore GL and Maranas CD. (2000) Modeling DNA mutation and recombination
for directed evolution experiments. J Theor Biol, 205(3): 483–503.
20. McCullum EO, Williams BA, et al. (2010) Random mutagenesis by error-prone PCR.
In: Braman J (editor) Methods Mol Biol New York: Humana Press. 634: 103–109.
21. Mondon P, Grand D, et al. (2010) Mutagen: a random mutagenesis method pro-
viding a complementary diversity generated by human error-prone DNA poly-merases. In: Braman J (editor) Methods Mol Biol New York: Humana Press. 634: 373–386.
22. Chiang LW, Kovari I, et al. (1993) Mutagenic oligonucleotide-directed PCR ampli-
fication (Mod-PCR): an efficient method for generating random base substitu-tion mutations in a DNA sequence element. PCR Methods Appl, 2(3): 210–217.
23. Lai YP, Huang J, et al. (2004) A new approach to random mutagenesis in vitro.
Biotechnol Bioeng, 86(6): 622–627.
24. Sambrook J and Russell DW, editors. (2001) Molecular Cloning: A Laboratory
Manual. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
25. Ausubel FM, Brent R, et al., editors. (2011) Current Protocols in Molecular Biol-
ogy John Wiley & Sons.
26. Saida F, Uzan M, et al. (2006) Expression of Highly Toxic Genes in E. coli: Special
Stragegies and Genetic Tools. Current Protein and Peptide Science, 7: 47–56.
Mutagenesis Application Guide
G
Gene construction 16, 20
Additions 7, 9, 10, 11, 23, 27, 31
Terminal additions 7, 9, 23, 27, 31
Oligonucleotide quality 22
Agarose gel 40, 41, 44, 45, 46
Reagents for mutagenesis 20
Annealing temperature 25, 26, 27, 39, 43, 44
Custom Mixed Bases 21
Antibiotics 48, 49
Ampicillin 48, 49
Machine Mixed Bases 21
Phosphate Modifications 20
Primers 20Trimers 21
Cassette mutagenesis. See Mutagenesis
Ultramer™ Oligonucleotides 20, 25
Cassettes 11, 16, 17
Universal bases 21
Chemical mutagenesis. See Mutagenesis
In vitro saturation. See Random mutagenesis
Cloning 5, 6, 12, 16, 20, 22, 23. See also Transfor-
Inverse PCR 6, 7, 12, 13, 14, 15
Competent cells 29, 30, 33, 36, 48, 49
Contamination 42, 49
Controls 29, 30, 41, 42, 47
KOD. See Polymerase
DpnI digestion controls 30, 47Ligation controls 30
PCR controls 29, 41, 42
Libraries 16, 18, 23
Transformation controls 30
Ligation 13, 14, 15, 30, 32, 44, 45, 46
Ligase 30, 32, 46
Dam methylase. See Methylated DNA
Melting temperature (Tm) 25, 26, 27, 28, 39, 42
Degenerate primers 21. See PCR with degenerate
Methylated DNA 12, 47
Deletions 6, 7, 9, 10, 11, 43, 46, 49
Mixed bases 19, 21
Denaturation temperature 44
DH5α. See Competent cells
Cassette mutagenesis 16, 17
Directed protein evolution 16
Chemical mutagenesis 19
DpnI. See Restriction enzyme
Random mutagenesis 5, 16, 19
Saturation mutagenesis 19Semi-random mutagenesis 19
E. coli. See Competent cells
Site-directed mutagenesis 5, 6, 7, 10, 12, 16, 22,
Electroporation 33, 36, 48
23, 24, 28, 39, 41, 43, 47
Enzymatic mutagenesis. See Error-prone PCR
Mutator strains 18
Error-prone PCR 6, 18Ethyl methane sulfonate (EMS) 19
N
Nucleotide analogs 18
Restriction enzyme 12, 16, 17, 26, 46
OligoAnalyzer 3.1 25, 26, 27, 39
DpnI 12, 24, 30, 32, 33, 36, 38, 47
Oligonucleotide-directed Internal Mutagenesis
Restriction site 6, 12, 16, 23, 26
23, 28, 31, 35, 42
Saturation mutagenesis. See Mutagenesis
PCR. See also Error-prone PCR; See also Inverse
SciTools® 25, 39. See also OligoAnalyzer 3.1; See
PCR; See also Touchdown PCR; See
also UNAFold
also Terminal additions by PCR; See
Screening for mutants 16, 29, 34, 37, 44
also Terminal changes by PCR
Semi-random mutagenesis. See Mutagenesis
Cycling conditions. See PCR Reaction param-
Sequencing 34, 37
Site-directed mutagenesis. See Mutagenesis
PCR with Degenerate Primers 19
Primer design 10, 25, 27, 28, 39
Reaction parameters 25, 29, 43
Substitutions 7, 8, 19
Phusion®. See Polymerase
Plasmid 12, 13, 14, 15, 16, 17, 28, 29, 30, 32, 34,
36, 37, 40, 41, 43, 44, 45, 46, 47, 49
T4 DNA Ligase 32. See also Ligation
Plasmid purification 34, 37, 44
Taq DNA polymerase. See Polymerase
Polymerase 12, 18, 19, 20, 21, 24, 31, 32, 35, 36,
Terminal additions by PCR 23, 27, 31
Terminal changes by PCR 23, 26, 31
High fidelity 12, 31, 32, 35, 36
KOD DNA Polymerase (Novagen) 24, 26, 31,
Toxic sequences 49
Transformation 30, 33, 36, 48
Pfu Turbo® (Stratagene) 35, 42
Phusion® (New England Biolabs) 31, 32, 35,
Ultramer™ Oligonucleotides 5, 6, 7, 9, 10, 16, 20,
Taq DNA polymerase 18, 32, 35, 42
Primer extension 6, 7, 10, 11, 12
Universal bases 21
Protocol for oligonucleotide-directed internal
UV irradiation 18
Protocol for terminal changes or additions 31
Vector 5, 16, 44, 46. See also Plasmid
Oligonucleotide 5, 7, 16, 20, 22, 25, 40, 41Template 40
Quick Ligation™ Kit (New England Biolabs) 32QuikChange® Site-Directed Mutagenesis Kit
(Stratagene) 12, 28, 39
R
Random mutagenesis. See Mutagenesis
Restriction digest 30, 32, 36, 38, 46, 47
Mutagenesis Application Guide
Ultramer™ and SciTools® are trademarks of Integrated DNA Technologies.
QuikChange® and Pfu Turbo® are registered trademarks of Stratagene Corporation.
GelStar® is a registered trademark of Cambrex Bio Science Rockland.
Phusion® and Quick Ligation™ are trademarks of New England Biolabs.
BigDye® is a registered trademark of Applied Biosystems.
Source: http://whitesci.co.za/wp-content/uploads/2011/12/Mutagenesis-Application-Guide.pdf
The ownership of U.S. industrial forestlands has dramatically changed since the 1980s. Whereas forest product companies had been the dominant owner of such lands and sent that timber to their mills, now the lands are owned by investment management organizations and trusts that manage the land to maximize returns. Some of these organizations, however, are delivering returns and helping to conserve the land.
Principal Idea: Estimating Survey 150 randomly selected students and 41% think marijuana should be legalized. Proportions If we report between 33% and 49% of all students at Chapter 10 the college think that marijuana should be legalized, how confident can we be that we are correct?


