
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Doi:10.1016/j.gene.2006.07.018

Gene 383 (2006) 43 – 51
A unique amino acid substitution, T126I, in human genotype C of hepatitis B
virus S gene and its possible influence on antigenic structural change
Fengrong Ren a,⁎, Asahito Tsubota b,d, Takatsugu Hirokawa a,c, Hiromitsu Kumada d,
Ziheng Yang e, Hiroshi Tanaka a
a Center for Information Medicine, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo, Tokyo 113-8510, Japan
b Institute of Clinical Medicine and Research, Jikei University School of Medicine, 163-1 Kashiwa-shita, Kashiwa, Chiba 277-8567, Japan
c Computational Biology Research Center, National Institute of Advanced Industrial Science and Technology, 2-41-6 Aomi, Koutou-ku, Tokyo 135-0064, Japan
d Department of Gastroenterology, Toranomon Hospital, 2-2-2, Toranomon, Minato, Tokyo 105-8410, Japan
e Department of Biology, University College London, Darwin Building, Gower Street, London WC1E 6BT, England, United Kingdom
Received 10 May 2006; received in revised form 21 June 2006; accepted 5 July 2006
Available online 29 July 2006
Received by Takashi Gojobori
Amino acid substitutions in the S gene of hepatitis B virus (HBV), especially in the ‘a' determinant region, have been suggested to affect the
antigenicity of the virus and the clinical outcome of the infected patient. However, no convincing evidence has been presented for this hypothesis,partly because the 3D structure of the S protein has not been determined. Comparative analysis of viral genes offers an approach to testing thishypothesis, as it may reveal signals of natural selection and provide insights into the functional significance of the observed amino acidsubstitutions. In this study, we analyze HBV S gene sequences obtained from 24 patients infected with HBV genotypes B or C, together with 16representative viral strains of HBV genotypes A–F retrieved from GenBank. We use phylogenetic methods to infer evolutionary changes amongHBV genotypes and to identify amino acid residues potentially under positive selective pressure. Furthermore, we employ the fragment assemblymethod to predict structural changes in the S protein. The results showed that an amino acid substitution within the ‘a' determinant, T126I, wasunique to genotype C, may affect the antigenicity of the HBsAg, and may result in poorer clinical outcomes of patients infected with genotype Cviral strains. We suggest that an integrated approach of evolutionary comparison and structural prediction is useful in generating hypotheses forfurther laboratory validation.
2006 Elsevier B.V. All rights reserved.
Keywords: Chronic HBV infection; Clinical outcome; ‘a' determinant; Ancestral viral sequence; Positive selection; 3D structure
with an increased risk of developing hepatocellular carcinoma(HCC), which is one of the major causes of human death.
The hepatitis B virus (HBV) has been well studied since the
HBV is a double-stranded DNA virus with a very compact
genome of only about 3200 bp. It encodes four proteins: S, P, C
). HBV infection, however,
and X. Some regions of the genome encode two proteins using
is still a significant worldwide public health problem. Chronic
different reading frames. The HBV has been divided into eight
HBV infection can lead to liver cirrhosis (LC), which severely
genotypes, A to H, based on an intergroup divergence of 8% or
damages liver function. Chronic HBV infection is also associated
greater of the complete nucleotide sequence, and these genotypesapparently have different geographic distributions (Recent studies haverevealed that there may be significant differences in clinical
Abbreviations: HBV, hepatitis B virus; 3D structure, three-dimensional
course and outcome among patients infected with different HBV
Corresponding author. Tel.: +81 3 58034762; fax: +81 3 58030247.
E-mail address: (F. Ren).
). For example, patients infected with
0378-1119/$ - see front matter 2006 Elsevier B.V. All rights reserved.
F. Ren et al. / Gene 383 (2006) 43–51
Fig. 1. Model of two-loop structure of the ‘a' determinant in the envelope gene of HBV. Small circles represent amino acids and bold lines represent the disulphidebridges. Two small solid circles represent the two substituted sites found in this study.
the genotype C virus were found to show poorer clinical outcomes
), called the ‘a' determinant, is located in the
than those infected with the genotype B virus, although both
central region (residues 124–147). The ‘a' determinant has been
genotypes are predominant in East Asia
predicted to be a double-loop structure projecting from the
). However, it is unclear which genetic differences
surface of the HBV particle (
between the genotypes are responsible for the clinical differences.
) (see and it has been suggested that the
One difficulty is the lack of sequential viral samples for
amino acid changes in this region could affect immune res-
longitudinal studies, which may be necessary for revealing
ponses (However, no convincing evidence at
evolutionary changes in the viral gene. Another difficulty is the
the 3D level has been presented for this hypothesis because of
lack of 3D structures of some HBV proteins, making it difficult to
the lack of information about the structure of the S protein.
assess the structural changes caused by amino acid substitutions
We employed bioinformatics approaches to infer amino acid
and their functional significance.
substitutions that probably have influenced the S protein structure
In this study, 24 HBV small surface antigen (HBsAg) se-
and so affected the HBsAg function. First, we performed
quences sampled from 24 patients showing quite different cli-
phylogenetic analysis using 24 sequences from patients as well
nical outcomes were analyzed. The HBsAg is the major
as representative strains of six HBV human genotypes, A to F,
component of the envelope of the hepatitis virion. It is 226
obtained from GenBank. We inferred the ancestral sequences of
amino acid residues long, completely embedded in the P gene
these viral strains with the reconstructed phylogenetic tree to
region (). A key region for HBV antigenicity
estimate what kind of amino acid substitutions occurred in each
Table 1Sequence number and basic clinical information of the 24 patients
Sequence Gender Date (age) of diagnosis of cirrhosis Genotype Subtype Development of HCC Development of SAE Clinical course
Alive (progressive)
Alive (progressive)
Alive (progressive)
Died of renal failure, GI bleeding
Died of hepatic failure
Died of pneumonia
Died of renal failure
Alive (progressive)
Died of hepatic failure
HCC: Hepatocellular carcinoma.
SAE: Severe acute exacerbation of chronic hepatitis accompanied by jaundice.
a Sequence in which two stop codons were found.
b Sequences in which one stop codon was found.
F. Ren et al. / Gene 383 (2006) 43–51
Fig. 2. The reconstructed phylogenetic tree of the 40 S gene sequences by the neighbor-joining method. Amino acid substitutions within the ‘a' determinant region ofthe S gene are shown along the branches, based on reconstruction of ancestral sequences using CODEML.
viral strain. We also detected sites that probably have undergone
nucleotide sequencing were obtained at the time when cirrhosis
positive selection using both S and P gene reading frames to
was confirmed by liver biopsy specimens, ultrasonography, and/
investigate the selection pressures acting on different genes.
or computed tomography. DNA extraction, polymerase chain
Second, the possible structural changes of the S protein caused by
reaction-based amplification, nucleotide sequencing and deter-
amino acid substitutions found in this study were computationally
mination of genotypes or subtypes were described previously
predicted at the 3D level. Finally, the possible relationship bet-
). All sequences determined were
ween amino acid substitutions and clinical outcomes was dis-
678 bp in length, without insertions or deletions in the alignment.
cussed based on the results obtained in this analysis.
Sixteen S gene sequences of genotypes A to F were retrieved
from GenBank and analyzed together with the 24 sequences
2. Materials and methods
determined in this study from Japanese patients. We selected thesesequences based on an evolutionary study of HBV in which each
2.1. Sequence data
of these viral strains was estimated to be representative of an HBVhuman genotype (They are
Twenty-four HBV S gene sequences were isolated from 24
HBVADW4A, HHVBF and HHVBFFOU for genotype F;
Japanese patients with chronic HBV infection, whose clinical
HHVBBAS and HHVBE4 for genotype E; HBVGEN1,
characteristics are shown in Serum samples analyzed for
HPBHBVAA and HBVAYWE for genotype D; HHVCCHA,
F. Ren et al. / Gene 383 (2006) 43–51
Fig. 3. Detection of positively selected sites. (a)–(b) show the results obtained by using the S gene reading frame, and (c)–(d) show the results by using the P genereading frame. The ordinate indicates the estimated probability for positively selected sites, whereas the abscissa indicates the amino acid site. The amino acid sites ofthe P gene are numbered from the starting position of the S gene, but two nucleotides are shifted.
HPBADR1CG and HPBCGADR for genotype C; HPBADW1,
Kimura's two-parameter model was used to calculate
HPBA3HMS2 and AB014366 for genotype B; and HBVGEN2
distances among the viral sequences, which are analyzed
and HVHEPB for genotype A. These HBV strains are included in
using the neighbor-joining method to
order to confirm the distribution of the 24 patient samples among
reconstruct the tree. To confirm whether or not the tree
the HBV phylogenies and also to infer the ancestral sequence of
topology depends on tree-making methods, we also used
each viral genotype. Sequences from genotypes G and H were not
maximum likelihood (to reconstruct the
used, as they are highly similar to those of genotypes A and F,
respectively A total of 40 S gene sequenceswere used in this study.
2.2.2. Inferring ancestral sequences
The amino acid sequences at the ancestral nodes of the
2.2. Phylogenetic analysis
reconstructed tree were inferred by using maximum likelihoodunder the JTT substitution model ), im-
2.2.1. Reconstructing the phylogenetic tree
plemented in the CODEML program in the PAML package
The phylogenetic relationships among the 40 strains were
Amino acid substitutions along
inferred using programs in the PHYLIP package
each branch were then examined.
F. Ren et al. / Gene 383 (2006) 43–51
2.3. Detection of positive selection pressure
The CODEML program was used to detect the presence of
3.1. Reconstructed phylogenetic tree
positively selected sites in the S protein. We used model8 (beta&ω) in the program, which assumes a mixture of sites
The reconstructed phylogenetic tree of the 40 HBV S genes
that are undergoing neutral evolution and that are under positive
is shown in The three genotype F strains were designated
selection (). We also conducted the same
as outgroups because the evidence of a major phylogenetic
analysis using the P gene reading frame because the region
division between genotypes A–E and genotype F has been
encoding the S gene overlaps with the P gene so that a sy-
shown by and also
nonymous substitution in the S gene may be a nonsynonymous
shows that the 40 sequences were clearly divided
substitution in the P gene.
corresponding to genotype classification, and its topology isalmost the same as that estimated by
2.4. Structure prediction of HBsAg
using non-overlapping regions. Seventeen samples of the 24patients were grouped into genotype B, and the remaining seven
To understand the functional influences of amino acid
samples were clustered into genotype C.
substitutions that occurred in the S gene, we employed com-putational prediction methods for investigating the 3D structure of
3.2. Estimated amino acid substitutions in ancestral sequences
the HBsAg. To choose an appropriate length for the prediction, wereferred to a recent study suggesting that residues 98 to 156 form a
To trace the evolutionary changes in the HBV virus,
multi-loop (MHL) structure that is especially hydrophilic
ancestral sequences at the nodes of the tree in were
). The MHL region may be important
inferred. Amino acid substitutions were found both upstream
to the conformation of the HBsAg structure, and so we used
and downstream of the sequences. Because of its significance to
residues 98 to 156 for the structure prediction.
antigenicity, we focus on the ‘a' determinant region. This region
The prediction was made using the fragment assembly
showed clear differences between genotypes. A T131N
method () on the ROBETTA protein structure
substitution (threonine to asparagine change at site 131) was
prediction server molecular mechanics (MM)/molecular dynamics (MD) calculation and a structural evaluation
program. Fragment assembly is very useful if a homology search
Positively selected sites with probability N0.5 estimated by CODEML
using BLAST (or PSI-BLAST
S gene reading frame
P gene reading frame
) fails to find a similar protein with a known structure.
16 representative
16 representative
In this case, the PSI-BLAST program did not detect any HBsAg
homologs in the Protein Data Bank (PDB,
for exploiting the template-based modeling approach. Therefore,
we employed the ROBETTA server in the first step to enumerate
the candidate models. This server automatically generates ten
models against a query sequence. In the second step, the most
similar topology model with the putative disulphide bridging
pattern derived from an amino acid mutation experiment
was selected from the 10 models by
human intervention. Three disulphide bond pairs in the model—
Cys124–Cys137, Cys107–Cys138 and Cys147–Cys139—could
be created via the connect bond command in MOE molecular
modeling software (Chemical Computing Group Inc.). Finally,
the model was refined using the MM/MD calculation on AMBER
8 ). The parm96 (force
field was used for all simulations. Three energy minimizations
were applied before the MD simulation: minimization of hy-
drogen atoms, side-chain minimization with the backbone
constraint and full atom minimization. The steepest descent and
conjugate gradient methods (maximum 10,000 cycles) were used
for each minimization. The 8 ns MD simulation was performed in
the NVT ensemble with the explicit water TIP3P model. The
temperature of the simulation was maintained at 300 K. The
Particle Mesh Ewald (PME) algorithm was used for electrostatic
interactions. The final model was selected from the last 4 ns based
on the structure quality score from the Verify3D protein structure
⁎: probability N0.95; ⁎⁎: probability N0.99.
evaluation program
Sites in the ‘a' determinant region of the S gene are in bold.

F. Ren et al. / Gene 383 (2006) 43–51
found on the branch leading to genotype A, a T126I substitution
the sequence. Within the ‘a' determinant region, only site 127
to genotype C and a F134Y substitution to genotype D. Also, a
was predicted to be a possible site for positive selection (with
T143S substitution was found on the branch that divided
probability P = 0.66) for the 16 representative HBV strains,
genotypes A and B from genotypes C–F. Since clinical
whereas two sites were detected in the samples of the 24
information is available for the 24 strains from Japanese
patients: site 126 (with P = 0.98) and site 143 (with P = 0.71).
patients, which are from genotypes B and C only, we are
On the other hand, the results obtained by using the P gene
especially interested in the branch connecting these two
reading frame showed a different pattern (–d). Seven
genotypes. Two amino acid changes were identified on that
sites were found to be under positive selection within the ‘a'
branch. The first is T126I, which is the third residue on the first
determinant for 16 representative strains, and four sites were
loop of the ‘a' determinant. The second is T143S, in the middle
detected in the samples of 24 sequences from the Japanese
of the second loop of the ‘a' determinant. Thus, except for C2,
patients, none of which had P N 0.95. Interestingly, the
all genotype C sequences of the patients have an isoleucine at
downstream region of the P gene seemed quite conserved
site 126 and, except for C1, all have a serine at site 143. In these
except for the last 20 residues for both the representative strains
two cases, a secondary substitution I126T occurred to C2 and
and the patient samples.
S143W to C1, respectively. In the genotype B group of thepatients, two of the 17 sequences were found to have amino acid
3.4. Predicted 3D structures of HBsAg
changes at site 126 as well. The first is a substitution T126I toB2, and the second is T126A to B7. Moreover, three sequences
shows the 3D structure of HBsAg predicted by using
were found to have stop codons in the 24 sequences from the
the fragment assembly method based on the B1 sequence,
Japanese patients: B6 with one, C2 with two, and C6 with one.
which is closest to the ancestor of genotype B. Ten models withdifferent fold topologies were initially obtained from the
3.3. Estimated positively selected sites of HBV S gene
ROBETTA prediction server based on fragment assemblymethod. Only one model had the putative three disulphide bond
and show the results of the codon-based
pairs consistent with the result that has been reported so far
analysis to identify positively selected sites. When the S gene
). This model then was refined using
reading frame was used (a–b), the central region seemed
MM/MD calculation and the feasibility of the model was
to be conserved compared to the upstream and downstream of
evaluated by Verify3D that calculates the structural quality
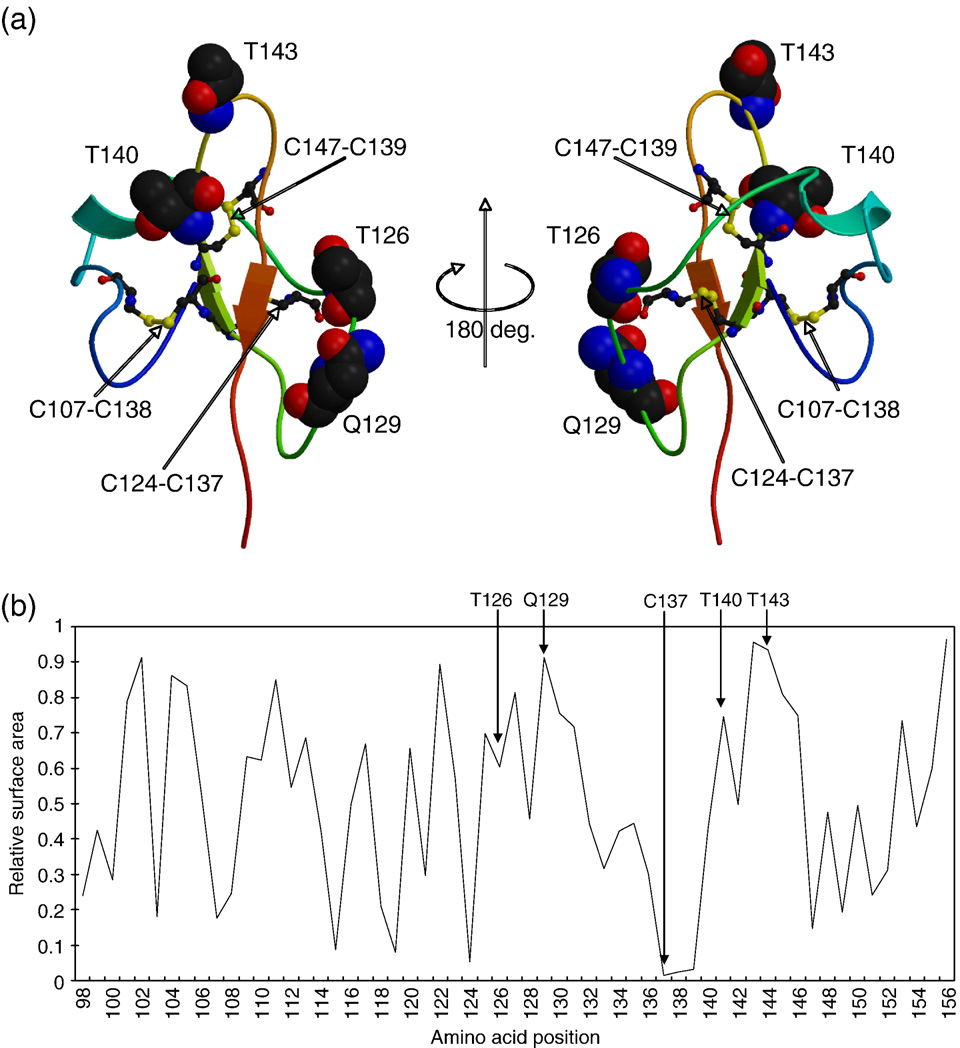
Fig. 4. (a) Cartoon representations of predicted 3D structure model of HBsAg (view facing the C-terminal region at left, rotated 180° at right about the y-axis). Severalkey substitution residues are shown in space filling representations. (b) A diagram of relative accessible surface area (RASA) of each amino acid position in the 3Dstructure model of HBsAg.
F. Ren et al. / Gene 383 (2006) 43–51
score. The best model from the last 4 ns MD trajectories had an
sequences from the Japanese patients. At site 143, in contrast,
acceptable structural quality score (25.44), which is fairly close
two amino acid substitutions, T143S and S143W, caused only
to the score expected for a correct structure (26.58) having this
minor changes in the hydropathy parameter (threonine: −0.7;
protein size. shows the best model with several key
serine: −0.8; tryptophan: −0.9).
substitution residues, which was prepared with MolScript
Third, patients whose S gene possessed an isoleucine at site
() and Raster3D We
126 exhibited poorer clinical conditions irrespective of geno-
also analyzed relative accessible surface area (RASA) of the
type classifications (see ). Therefore, it seemed rea-
residues in this model (b) using InsightII/Homology
sonable to suppose that the T126I substitution has a major
program (Accelrys Inc.). It was estimated that four sites–T126,
impact on the antigenicity of HBV.
Q129, T140 and T143–were exposed on the surface of HBsAg.
Predicted 3D structure. Our hypothesis is supported by the
results of the structure prediction of HBsAg. As mentioned
above, the B1 sequence is the closest to the ancestral branch ofgenotype B in the phylogenetic tree, and no amino acid
Amino acid substitutions in the HBV S gene, especially in
substitution occurred in the ‘a' determinant to this sequence.
the ‘a' determinant region, have been described in vaccinated
Thus it is appropriate to discuss the structural difference
children and patients treated with hepatitis B immunoglobulin.
between genotypes B and C based on the predicted result of this
Many studies have pointed out that replacement at some amino
sequence. Four amino acid sites–126, 129, 140 and 143–were
acid sites could affect the antigenicity of the HBsAg, resulting
predicted to be exposed to the surface of HBsAg (). As the
in the loss of recognition by antibodies and leading to evasion of
T126I substitution involves the largest change in chemical
the virus from the neutralizing antibody response (
properties, it is most likely to cause structural changes in the
Since most reported amino acid substitutions have been
Stop codons. Stop codons are usually not allowed in protein-
found within the second of the two loops of the ‘a' determinant,
coding genes, as they cause the unexpected termination of
the second loop has been considered important; in particular,
protein translation. However, stop codons were found in the
sites 141 to 145 are thought to be essential for antibody binding
PreC/C encoding region, and they resulted in immunological
escape of the virus and led to fulminant hepatitis
We focus on the 24 sequences from the Japanese patients, as
). Stop codons observed in the S gene in the samples of 24
clinical information is available for them only. Out of the 10
Japanese patients may have a similar role, as all patients with
amino acid substitutions inferred to have occurred in the sample,
stop codons in the viral S gene showed worse clinical outcomes
7 are found in the ‘a' determinant region Since patients
irrespective of the genotype.
infected with the genotype C virus had poorer clinical outcomes
Positively selected sites. The amino acid sequence in the ‘a'
than those infected with the genotype B virus, we focus on the
determinant is thought to be highly conserved due to its bio-
ancestral branch that separates genotypes B and C. Two amino
logical function, with substitutions being restricted to similar
acid substitutions were observed on these branches: T126I and
amino acids (). In our analysis, the ‘a' deter-
minant region was conserved on the whole, but site 126 was
T126I—unique substitution to genotype C. We suggest that
inferred to be under positive selection with high probability
the T126I substitution in the first loop may be more important
(0.98). Thus, amino acid substitutions at this site might lead to
than the T143S substitution in the second loop. First, the T126I
adaptive evolution of the viral gene, resulting in altered anti-
substitution was found on the ancestral branch that directly
genicity and increased virulence. In contrast, a conserved region
leads to the genotype C group on the reconstructed tree
was detected to be located in the downstream rather than in the
The results of a study on the genetic diversity of HBV by
‘a' determinant when the P gene reading frame was used, which
support this finding. They analyzed 630
obviously corresponds to the distribution of the functional
HBV S gene sequences, including all human genotypes as well
domains of the P gene.
as nonhuman primate HBV strains (chimpanzee, gorilla, gibbon
The P gene has been divided into several functional regions. The
and orangutan) and found that most human genotype C strains
DNA polymerase region overlaps with the S gene
have 126I, whereas almost all human nongenotype C strains
). Within the polymerase domain, so-called A–E regions can
have 126T. On the other hand, T143S occurred on the branch
be identified on the basis of homology with corresponding regions
that separated genotype B and all other genotypes, and thus it
of other polymerases. It has been reported that the C region
was not unique to genotype C.
(residues 547–559) in which a tyrosine–methionine–aspartate–
Second, the large difference in chemical properties between
aspartate (YMDD) motif (residues 551–554) exists is especially
threonine and isoleucine means that the T126I substitution may
important in function and thus is strictly conserved (
have a major impact on the antigenicity of the HBsAg.
Apparently, the coding regions for important functions of
According to method for displaying
these two genes do not overlap. In addition, a familiar
the hydropathy parameter of a protein, isoleucine has the
substitution, YMDD to YIDD, which can affect the outcome of
highest value (+ 4.5) among 20 amino acids, while threonine
chronic hepatitis, was not found in any of the 16 representative
shows a negative value (−0.7). This is the largest difference
strains or 24 patient sequences (Thus the effect from this
among amino acid replacements observed in the samples of 24
substitution can be excluded in this study.
F. Ren et al. / Gene 383 (2006) 43–51
Fig. 5. Aligned partial sequences of the P gene at amino acid level by ClustalW. (a) is the result of 16 representative HBV strains from the database, whereas (b) is thatof the 24 patient sequences. The YMDD motif is indicated by a light grey shadow.
Overlapping reading frames in the viral genome. The over-
study may also influence clinical outcomes, including the viral
lapping reading frames in the viral genome may cause difficulty
subtype and the patient's age ).
in codon-based analysis, because a nonsynonymous substitu-
Finally, we would like to emphasize that combining molecular
tion in one gene might be a synonymous substitution in another.
evolutionary analysis with protein structure prediction appears to
However, the functionally important regions of the S and P
be a powerful approach to the study of viral evolution. The
genes appear to be separate in the HBV genome. The portion of
evolutionary analysis can pinpoint when and where important
the P gene that overlaps with the S gene does not appear to be
amino acid substitutions occurred, and structural prediction helps
functionally important except for the conserved YMDD motif,
to assess their functional significance. The combined approach
so the evolutionary process of this region of the genome is
appears to be effective in generating biological hypotheses, which
dominated by the selective pressure on the S gene. Our results
may be verified through further experimental tests.
detecting positively selected sites in the S gene are also highlyconsistent with the predicted structural changes in the S protein.
The results of this study suggest that evolutionary changes in
the S gene may be important in determining the clinical outcome
This study is supported by a Grant-in-Aid for Scientific
of a hepatitis B patient. Important changes include substitutions
Research from the Ministry of Education, Culture, Sports,
that drastically change the properties of amino acids in key regions
Science and Technology of Japan to F.R. and H.T.
of the protein, such as the ‘a' determinant, and substitutions to stopcodons. The unique amino acid change, T126I, observed in
genotype C, is strongly suspected to be one of the factors thataffect the antigenicity of the HBsAg and to result in poorer clinical
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., 1990. Basic
outcomes. It should be noted that factors not considered in this
local alignment search tool. J. Mol. Biol. 215, 403–410.
F. Ren et al. / Gene 383 (2006) 43–51
Altschul, S.F., et al., 1997. Gapped BLAST and PSI-BLAST: a new generation
Norder, H., Hammas, B., Lofdahl, S., Courouce, A.M., Magnius, L.O., 1992.
of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Comparison of the amino acid sequences of nine different serotypes of
Berman, H.M., et al., 2000. The protein data bank. Nucleic Acids Res. 28, 235–242.
hepatitis B surface antigen and genomic classification of the corresponding
Blumberg, B.S., Alter, H.J., Visnich, S., 1965. A new antigen in leukemia sear.
hepatitis B virus strains. J. Gen. Virol. 73, 1201–1208.
JAMA 191, 541–546.
Norder, H., et al., 1993. Genetic relatedness of hepatitis B viral strains of diverse
Chu, C.J., Hussain, M., Lok, A.S., 2002. Hepatitis B virus genotype B is
geographical origin and natural variations in the primary structure of the
associated with earlier HBeAg seroconversion compared with hepatitis B
surface antigen. J. Gen. Virol. 74, 1627–1632.
virus genotype C. Gastroenterology 122, 1756–1762.
Norder, H., Courouce, A.M., Magnius, L.O., 1994. Complete genomes,
Cornell, W.D., et al., 1995. A second generation force field for the simulation of
phylogenetic relatedness, and structure proteins of six strains of the hepatitis
proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117,
B virus, four of which represent two new genotypes. Virology 198, 489–503.
Norder, H., et al., 2004. Genetic diversity of hepatitis B virus strains derived
Dane, D.S., Cameron, C.H., Briggs, M., 1970. Virus-like particles in serum of
worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology
patients with Australia-antigen-associated hepatitis. Lancet i, 695–698.
47, 289–309.
Ding, X., et al., 2001. Hepatitis B virus genotype distribution among chronic
Okamoto, H., et al., 1988. Typing hepatitis B virus by homology in nucleotide
hepatitis B virus carriers in Shanghai, China. Intervirology 44, 43–47.
sequence: comparison of surface antigen subtypes. J. Gen. Virol. 69, 2575–2583.
Fares, M., Holmes, E., 2002. A revised evolutionary history of hepatitis B virus
Okochi, K., Murakami, S., 1968. Observation on Australia antigen in Japan. Vox
(HBV). J. Mol. Evol. 54, 807–814.
Sang. 15, 374–385.
Felsenstein, J., 1981. Evolutionary trees from DNA sequences: a maximum
Orito, E., et al., 2001a. A case-control study for clinical and molecular biological
likelihood approach. J. Mol. Evol. 17, 368–376.
differences between hepatitis B viruses of genotypes B and C. Japan HBV
Felsenstein, J., 1995. PHYLIP: Phylogeny Inference Package, Ver. 3.572.
Genotype Research Group. Hepatology 33, 218–223.
University of Washington, Seattle, WA.
Orito, E., et al., 2001b. Geographic distribution of hepatitis B virus (HBV)
Fujii, H., et al., 1992. Gly 145 to Arg substitution in HBsAg antigen of immune
genotype in patients with chronic HBV infection in Japan. J. Viral Hepatitis
escape mutant of hepatitis B virus. Biochem. Biophys. Res. Commun. 184,
34, 590–594.
Pearlman, D.A., et al., 1995. AMBER, a package of computer programs for
Galibert, F., Madart, E., Fitoussi, F., Tiollais, P., Charnay, P., 1979. Nucleotide
applying molecular mechanics, normal mode analysis, molecular dynamics
sequences of the hepatitis B virus genome (subtype ayw) cloned in E. coli.
and free energy calculations to simulate the structural and energetic
Nature 281, 646–650.
properties of molecules. Comput. Phys. Commun. 91, 1–41.
Ghany, M.G., et al., 1998. Hepatitis B virus S mutants in liver transplant
Protzer-Knolle, U., et al., 1998. Hepatitis B virus with antigenically altered
recipients who were reinfected despite hepatitis B immune globulin
hepatitis B surface antigen is selected by high-dose hepatitis B immune
prophylaxis. Hepatology 27 (1), 213–222.
globulin after liver transplantation. Hepatology 27, 254–263.
Grandjacques, C., et al., 2000. Rapid detection of genotypes and mutations in the
Saitou, N., Nei, M., 1987. The neighbor-joining method: a new method for
pre-core promoter and the pre-core region of hepatitis B virus genome:
reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
correlation with viral persistence and disease severity. J. Hepatol. 33, 430–439.
Simons, K.T., Kooperberg, C., Huang, E., Baker, D., 1997. Assembly of protein
Gunther, S., et al., 1999. Absence of mutations in the YMDD motif/B region of
tertiary structures from fragments with similar local sequences using simulate
the hepatitis B virus polymerase in famciclovir therapy failure. J. Hepatol.
annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225.
30, 749–754.
Stirk, H.J., Thornton, J.M., Howard, C.R., 1992. A topological model for
Howard, C.R., 1995. The structure of hepatitis B envelope and molecular
hepatitis B surface antigen. Intervirology 33, 148–158.
variants of hepatitis virus. J. Viral Hepatitis 2, 165–170.
Tiollais, P., Charnay, P., Vyas, G.N., 1981. Biology of hepatitis. Science 213,
Jones, D.T., Taylor, W.R., Thornton, J.M., 1992. The rapid generation of mutation
data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282.
Tiollais, P., Pourcel, C., Dejean, A., 1985. The hepatitis B virus. Nature 317,
Kao, J., Chen, P., Lai, M., Chen, D., 2000. Hepatitis B genotype correlates with
clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118,
Tsubota, A., et al., 1998. Deletions in the hepatitis B virus core gene may
influence the clinical outcome in hepatitis B e antigen-positive asymptom-
Kim, D.E., Chivian, D., Baker, D., 2004. Protein structure prediction and
atic healthy carriers. J. Med. Virol. 56 (4), 287–293.
analysis using the Robetta server. Nucleic Acids Res. 32, 526–531.
Tsubota, A., Arase, Y., Ren, F., Tanaka, H., Ikeda, K., Kumada, H., 2001.
Kosaka, Y., et al., 1991. Fulminant hepatitis B: induction by hepatitis B virus
Genotype may correlate with liver carcinogenesis and tumor characteristics
mutants defective in the precore region and incapable of encoding e antigen.
in cirrhotic patients infected with hepatitis B virus subtype adw. J. Med.
Gastroenterology 100, 1087–1094.
Virol. 65, 257–265.
Kraulis, P.J., 1991. MOLSCRIPT: a program to produce both detailed and
Weinberger, K.M., Bauer, T., Bohm, S., Jilg, W., 2000. High genetic variability of
schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–950.
the group-specific a-determinant of hepatitis B virus surface antigen (HBsAg)
Kyte, J., Doolittle, R.F., 1982. A simple method for displaying the hydropathic
and the corresponding fragment of the viral polymerase in chronic virus carriers
character of a protein. J. Mol. Biol. 157 (1), 105–132.
lacking detectable HBsAg in serum. J. Gen. Virol. 81, 1165–1174.
Luthy, R., Bowie, J.U., Eisenberg, D., 1992. Assessment of protein models with
Yang, Z., 1997. PAML: a program package for phylogenetic analysis by
three-dimensional profiles. Nature 356, 83–85.
maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (
Mayerat, C., Mantegani, A., Frei, P.C., 1999. Does hepatitis B virus (HBV)
genotype influence the clinical outcome of HBV infection? J. Viral Hepatitis
Yang, Z., Kumar, S., Nei, M., 1995. A new method of inference of ancestral
6, 299–304.
nucleotide and amino acid sequences. Genetics 141, 1641–1650.
Merritt, E.A., Bacon, D.J., 1997. Raster3D: photorealistic molecular graphics.
Yang, Z., Nielsen, R., Goldman, N., Pedersen, A.K., 2000. Codon-substitution
Methods Enzymol. 277, 505–524.
models for heterogeneous selection pressure at amino acid sites. Genetics
Ni, F., Fag, D., Gan, R.B., Li, Z.P., 1995. A new escape mutant of hepatitis B
155, 431–449.
virus with an Asp to Ala substitution in aa 144 of the envelope major protein.
Res. Virol. 146 (6), 205–210.
Source: http://abacus.gene.ucl.ac.uk/ziheng/pdf/2006RenGenev383p43.pdf
Nacho is fast becoming Nacho again, despite chronic kidney disease Find out how your cat's kidney disease can be managed with Semintra Finding out your cat has chronic kidney chronic kidney disease in cats After many years together, finding Many owners are aware that cats can
NATIONAL LIFE SCIENCES P2 VERSION 1 (NEW CONTENT) FOR FULL-TIME CANDIDATES NOVEMBER 2011 MARKS: 150 This memorandum consists of 11 pages. Copyright reserved Please turn over Life Sciences/P2 (Version 1) (Full-time) DBE/November 2011 NSC – Memorandum PRINCIPLES RELATED TO MARKING LIFE SCIENCES 2011


