
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
06-3344 1626.1635
The Human Papillomavirus E6 Oncogene Dysregulates the CellCycle and Contributes to Cervical Carcinogenesis throughTwo Independent Activities
Anny Shai,1 Tiffany Brake,1 Chamorro Somoza,2 and Paul F. Lambert1
1McArdle Laboratory for Cancer Research, University of Wisconsin School of Medicine and Public Health,Madison, Wisconsin and 2Arbor Vita Corporation, Sunnyvale, California
we generated K14E6WT and K14E7WT transgenic mice expressingeither the HPV16 E6 or the HPV16 E7 oncogene, respectively.
Cervical cancer is a leading cause of death due to cancer
The human keratin-14 promoter was used to direct transgene
among women worldwide. Using transgenic mice to dissect the
expression to the basal layer of the stratified squamous epithelium
contributions of the human papillomavirus (HPV) 16 E6 and
lining the skin, oral cavity, and reproductive tract (5–7). K14E6WT
E7 oncogenes in cervical cancer, E7 was identified previously
and K14E7WT transgenic mice display many of the known activities
to be the dominant oncogene. Specifically, when treated with
of each oncogene identified in tissue culture, including the ability
exogenous estrogen for 6 months, E7 transgenic mice
of E6 and E7 to inactivate p53 and pRb, respectively. Furthermore,
developed cancer throughout the reproductive tract, but E6
these HPV16 transgenic mice develop tumors in the skin either
transgenic mice did not. E6 contributed to carcinogenesis of
spontaneously or with increased efficiency when induced chemi-
the reproductive tract, as E6/E7 double transgenic mice
cally with the carcinogens, 7,12-dimethylbenz(
treated for 6 months with estrogen developed larger cancers
than E7 transgenic mice. In the current study, we investigated
O-tetradecanoylphorbol-13-acetate (5, 6, 8).
Prior studies showed that both the K14E6WT/K14E7WT trans-
whether the E6 oncogene alone could cooperate with estrogento induce cervical cancer after an extended estrogen treat-
genic mice and the K14E7WT singly transgenic mice developed
ment period of 9 months. We found that the E6 oncogene
cervical cancer following estrogen treatment for 6 months.
synergizes with estrogen to induce cervical cancer after
However, similarly treated K14E6WT transgenic mice developed
9 months, indicating that E6 has a weaker but detectable
only low-grade dysplasia (7). Estrogen is a cofactor in cervical
oncogenic potential in the reproductive tract compared with
carcinogenesis in this mouse model, as untreated K14E6WT/
the E7 oncogene. Using transgenic mice that express mutant
K14E7WT or K14E7WT mice did not develop cancer. Subsequent
forms of HPV16 E6, we determined that the interactions of E6
studies indicated that estrogen is required for multiple stages of
with cellular A-helix and PDZ partners correlate with its
cervical carcinogenesis (9). Reproductive tumors arising in the
ability to induce cervical carcinogenesis. In analyzing the
K14E6WT/K14E7WT transgenic mice were more aggressive than
tumors arising in E6 transgenic mice, we learned that E6
those arising in the K14E7WT transgenic mice, indicating that the
induces expression of the E2F-responsive genes, Mcm7 and
E6 oncogene contributed to the malignant progression.
cyclin E, in the absence of the E7 oncogene. E6 also prevented
In the current study, we investigated whether the E6 oncogene
the expression of p16 in tumors of the reproductive tract
could cooperate with estrogen to induce cervical cancer given an
through a mechanism mediated by the interaction of E6 with
extended (9 months) treatment period. To examine the mecha-
A-helix partners. [Cancer Res 2007;67(4):1626–35]
nism(s) by which the E6 oncogene contributes to cervical can-cer, we monitored cervical carcinogenesis in K14E6I128T andK14E6D146-151 mice, which express mutant forms of the HPV16 E6
oncogene (10, 11). K14E6I128T transgenic mice express a mutant
Cervical cancer is the second most common type of cancer
form of E6 greatly reduced in its ability to bind a-helix partners.
among women, with high mortality rates worldwide, despite
Specifically, E6I128T protein binds the a-helix partners, E6AP and
increased screening efforts (1). Human papillomavirus (HPV)
E6BP, at 1% to 5% the levels of wild-type (WT) E6 protein (12).
infection contributes to nearly all cases of cervical cancer based
E6AP or UBE3A belongs to the HECT family of E3 ubiquitin ligases
on the observed presence of HPV DNA within these cancers (2) and
(13) and is normally associated with the human neurologic
more than half of the HPV-associated cervical cancers are
disorder, Angelman's syndrome (14, 15). E6AP is thought to be
attributed to infection with HPV16 (2, 3).
the primary cellular factor mediating the degradation of p53 by E6
Two viral genes, E6 and E7, are commonly expressed in cervical
(16) and is thus a potentially important partner in mediating the
cancer. In tissue culture, E6 and E7 display properties of
oncogenic activities of E6. Correspondingly, K14E6I128T transgenic
oncogenes, including the ability to immortalize and transform
mice are defective for inactivating p53 (17). K14E6D146-151 trans-
cells (4). To assess the oncogenic properties of these genes in vivo,
genic mice encode a mutant E6 protein defective for interactingwith PDZ partners (18), such as DLG and Scribble, two genesknown to be tumor suppressors in Drosophila. We have previouslyused K14E6I128T and K14E6D146-151 transgenic mice to show a role
Requests for reprints: Paul F. Lambert, McArdle Laboratory for Cancer Research,
of the a-helix and PDZ domain partners of E6, respectively, in
University of Wisconsin School of Medicine and Public Health, Madison, WI 53706.
mediating the oncogenic potential of E6 in the skin (10, 17, 19).
Phone: 608-262-8533; Fax: 608-262-2824; E-mail:
[email protected].
These studies provide a framework for our studies of cervical
I2007 American Association for Cancer Research.
doi:10.1158/0008-5472.CAN-06-3344
carcinogenesis studies reported herein.
Cancer Res 2007; 67: (4). February 15, 2007
Multiple Properties of E6 Contribute to Cervical Cancer
We found that the E6 oncogene synergizes with estrogen to induce
lesion scored as the final diagnosis. The tumors were measured with the
cervical cancer after an extended estrogen treatment. Specifically,
Zeiss Axiovision (version 3.1) program (Zeiss, Thorwood, NY). Any tumor
K14E6WT mice, treated with estrogen for 9 months, developed
with an area >2,000 Am2 was classified as a large cancer.
cervical cancers at an increased frequency compared with non-
Quantification of bromodeoxyuridine. To quantify the amount of
basal DNA synthesis, the total number of bromodeoxyuridine (BrdUrd)–
transgenic mice. Compared with K14E6WT mice, K14E6I128T and
positive basal cells was counted and divided by the total number of basal
K14E6D146-151 mice in the absence and/or the presence of E7
cells and multiplied by 100 to determine the percentage. To quantify the
displayed reduced oncogenic potential in the reproductive tract.
amount of epithelial hyperplasia, the total number of suprabasal BrdUrd-
We also evaluated mouse reproductive tracts and their
positive cells were counted and divided by the total number of cells and
associated tumors for biomarkers commonly used for the
multiplied by 100 to determine the percentage. BrdUrd was counted in
diagnosis of human cervical cancers. Biomarkers included the
eight, !40 microscopic fields per mouse, with a total of at least three or
E2F-responsive genes Mcm7, involved in DNA replication (20),
more mice per genotype group.
and cyclin E, involved in the G
Statistical analysis. The Fisher's exact test was used to determine the
1-S transition (21, 22). We also
monitored expression of the cyclin kinase inhibitor, p16. p16 is a
significance in tumor incidence. The Wilcoxon rank-sum test was used to
biomarker for HPV-associated cervical lesions and cancers (23).
determine the significance in BrdUrd quantification and in measurementsof tumor size and area. Statistical analysis was carried out using the MSTAT
Finally, tumors were evaluated for p53 expression. In most cases,
biomarker expression in the lesions and tumors from our
Immunohistochemistry. Sections were prepared for immunohisto-
nontransgenic and transgenic mice mirrored results observed in
chemical analysis as described previously (7). For BrdUrd, cyclin E, pRb, and
human cervical samples. Of particular interest, lesions from our
p16 analysis, the slides were immersed in 2N HCl for 20 min to unmask
K14E6WT and K14E6mutant (refers to both K14E6I128T and
further. Primary antibody was applied to the sections at 1:100 for BrdUrd
K14E6D146-151 herein) mice showed an up-regulation of the
(Calbiochem, Darmstadt, Germany); 1:200 to 1:500 in blocking solution for
E2F-responsive genes, Mcm7 and cyclin E, even in the absence
p53 (CM5; Novocastra, Norwell, PA); and 1:50 for MCM7 (NeoMarkers,
of E7, but at lower levels than in tumors arising in E7-expressing
Fremont, CA), cyclin E (M-20; Santa Cruz Biotechnology, Santa Cruz, CA),
mice. In contrast, we observed an inverse relationship between
pRb (BD Biosciences, San Jose, CA), and p16 (M156; Santa Cruz
the expression of p16 and pRb in the reproductive tumors from
Biotechnology) overnight at 4jC. A universal biotinylated secondaryantibody was applied and developed.
K14E6WT versus K14E7WT mice. Biomarker expression in tumorsarising in K14E6WT/K14E7WT mice were similar to tumors arisingin K14E7WT mice, showing that E7 is the dominant oncogene in
deregulating the p16/pRb pathway during cervical carcinogenesis.
K14E6WT and K14E6mutant transgenic mice express physio-
In summary, this study shows that two properties of E6
logic levels of E6 protein in the epithelia of the skin and
contribute to the development of cervical cancer. These
reproductive tract. Prior detection of HPV16 E6 protein was
contributions lead to a distinct pattern of dysregulation of cell
difficult due to an absence of adequately sensitive antibodies. A
cycle regulatory genes compared with that seen in E7-expressing
recently generated HPV16 E6–specific antibody (24) allowed us to
detect and compare levels of E6 protein expressed in ourtransgenic mice with cell lines derived from human precancerous
Materials and Methods
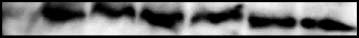
cervical intraepithelial neoplasia (CIN) lesions and cervical cancers(Fig. 1). HPV16 E6 protein was detected in the HPV16-positive SiHa
Mouse lines and estrogen treatment. The K14E6WT (5), K14E6I128T
and Caski but not in the HPV18-positive HeLa and HPV-negative
(17), K14E6D146-151 (10), and K14E7WT (6) lines were all maintained on aheterozygous FVB background. K14E6WT, K14E6I128T, and K14E6D146-151
C33a cervical cancer cells (Fig. 1A). E6 protein was also detected in
mice were mated to K14E7WT mice to generate double transgenic mice.
W12 clonal cell lines derived from a HPV16-positive CIN1 lesion.
Female mice were treated with 17-h estradiol as described previously (7)
Dorsal skin from 9-day-old WT and mutant E6 transgenic mice
for 9 months. Untreated control mice were held for the same period. All
expressed E6 protein at levels slightly higher than SiHa cells,
mice were bred and maintained in the American Association for
whereas expression in adult mice were lower (Fig. 1B; data not
Accreditation of Laboratory Animal Care–approved McArdle Laboratory
shown). This result is consistent with our prior observations that
Animal Care Facility in accordance with an institutionally approved
K14-directed transgene expression is maximal in young pups and
animal protocol.
wanes in adults (25). In homozygous K14E6WT transgenic mice, the
Quantification of E6 levels. Five-week-old female mice were treated
level of E6 protein was approximately half of the amount of E6
with estrogen, and reproductive tracts were harvested after 6 weeks of
expressed in Caski cells (Fig. 1B). E6 protein was detected in the
treatment to obtain a state of constant estrus in all of the mice. Dorsal skinwas also harvested at the time of sacrifice in addition to skin from 9-day-old
reproductive tracts of all adult K14E6WT and K14E6mutant mice, at
mice, as transgene expression is highest during this period in the skin. The
levels lower than in both SiHa and Caski cells (Fig. 1C). Given that
tissue was placed in cold HNTG lysis buffer [50 mmol/L HEPES (pH 7.5),
<10% of the total protein from the harvested reproductive tract
150 mmol/L NaCl, 1.1% Triton X-100, 1 mmol/L EGTA, 10% glycerol,
tissues comes from the stratified epithelium, we conclude that our
1 mmol/L phenylmethylsulfonyl fluoride, 1! PIC] and homogenized.
K14E6WT transgenic mice express E6 protein at levels similar to
Protein lysates (100–200 Ag) were separated by SDS-PAGE, transferred,
that observed in human cervical cancer. K14E6D146-151 mice
and immunoblotted with a monoclonal HPV16 E6 antibody (24) at 5 Ag/mL.
expressed f1.5 times the amount of E6 protein relative to
A mouse IgG secondary conjugated to horseradish peroxidase (Jackson
K14E6WT mice in the reproductive epithelium (Fig. 1C). K14E6I128T
Immunoresearch, West Grove, PA) was used at 1:10,000. Detection of E6 was
transgenic mice (data not shown) expressed mutant E6 protein
achieved by using the Enhanced Chemiluminescence Plus Western
roughly equal to that in K14E6WT mice.
Detection kit (Amersham, Piscataway, NJ).
Analysis of reproductive tracts. Reproductive tracts were harvested
after 9 months of estrogen treatment and analyzed as described previously(7). The fixed tissue was histologically sectioned and every tenth 5-Amsection was stained with H&E and pathologically examined, with the worst
Cancer Res 2007; 67: (4). February 15, 2007







E6 synergizes with estrogen in the absence of E7 to form
reproductive tumors after estrogen treatment for 9 months.
Whereas K14E6WT transgenic mice did not develop cancer after6 months of estrogen treatment, E6 contributed to the severity oftumors arising in K14E6WT/K14E7WT mice when treated withestrogen for either 6 or 9 months (7, 9). In the current study,K14E6WT mice were treated with estrogen for an extended 9-monthperiod to investigate whether E6 could induce cervical cancer inthe absence of E7. As expected, none of the untreated mice,regardless of genotype, developed cancer (data not shown). After9 months of estrogen treatment, 41% of K14E6WT mice developedtumors in the reproductive tract compared with 6.7% fornontransgenic mice (Table 1). This difference was statisticallysignificant (P = 0.02). In contrast, 100% of K14E7WT transgenic micetreated for 9 months developed cancer (9). Thus, E6, in the absenceof E7, can induce cervical cancers in cooperation with exogenousestrogen, albeit less robustly than E7.
An E6 mutant reduced in binding A-helix partners has a
lower incidence of cancer and develops smaller tumors. E6binds to numerous cellular proteins. One subset (e.g., the E3ubiquitin ligase E6AP) binds to E6 via a leucine-rich a-helical(a-helix) motif, whereas another (e.g., Dlg and Scribble) bindsthrough PDZ domains. We used two lines of E6 mutant mice,K14E6I128T and K14E6D146-151, defective in binding a-helix and PDZpartners, respectively (10, 17), to examine the importance of theinteraction of E6 with each subset of partners in mediatingthe oncogenic properties of E6 in the reproductive tract. After a9-month treatment period with estrogen, K14E6I128T mice had amarginally significant reduction in tumor incidence relative toK14E6WT mice (19.4% versus 41%; P = 0.058; Table 1). In contrast,the K14E6D146-151 transgenic mice had similar rates of cancerincidence as the K14E6WT mice (P = 0.25). Tumors from thereproductive tract of K14E6WT and K14E6mutant mice were variablein size (Fig. 2A). Nonetheless, tumors arising in the K14E6I128Ttransgenic mice were generally smaller relative to tumors fromeither K14E6WT or K14E6D146-151 mice (Table 1). The largest tumorsize on average for K14E6I128T mice (1.38 mm2 cross-sectional area)was significantly smaller (P = 0.041) than that for K14E6WT mice(5.22 mm2). In contrast, there was no significant difference in thelargest tumor size for the K14E6D146-151 mice compared with the
Figure 1. Comparison of E6 expression in K14E6 transgenic mice andhuman cervical cancer cell lines. A, levels of E6 in HPV-positive andHPV-negative cervical cell lines. Immunoblots probed with antibodies specificfor either E6 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The GAPDH-specific immunoblot was done to confirm equivalence in loading
Table 1. Comparison of tumors from the reproductive
and was done for all experiments displayed in this figure, although shownonly for the top-most one. For each sample, 200 Ag of total soluble protein
tract of nontransgenic, K14E6WT and K14E6mutant
were analyzed. The top blot was loaded with samples from HPV-negative
C33A, HPV18-positive HeLa, and HPV16-positive SiHa and Caski humancervical cancer–derived cell lines. The second blot was loaded with samplesfrom various clonal populations of W12E (20850 and 20963) and W12I
(20861, 20822, 20862, and 201402) cell lines derived from a HPV16-positive
incidence (%) multiplicity tumor (mm2)
CINI lesion. W12E clones harbor the viral genome in the extrachromosomalstate. W12I clones harbor the genome in a chromosomally integrated state.
B, levels of E6 in the dorsal skin of K14E6WT and K14E6mutant transgenic
mice at age postnatal day 9 or 10. In this immunoblot, 200 Ag (top) or
250 Ag (bottom) of total cellular protein from each mouse tissue sample and
K14E6I128T (n = 36)
100 or 125 Ag of SiHa and Caski extracts were analyzed. Extracts from
K14E6D146-151 (n = 28)
different animals (A and B) of the same genotype were loaded to assessreproducibility of findings. Bottom, levels of E6 protein in E6 homozygoustransgenic mice in both skin and ear. C, levels of E6 in the reproductive tractof K14E6WT and K14E6mutant transgenic mice. Top, 200 Ag of total cellular
Abbreviation: NTG, nontransgenic.
protein from each mouse tissue sample were analyzed. As above, extracts
*Cancer incidence in K14E6I128T was marginally significant compared
from different animals (A, B , and C ) of the same genotype were loaded toassess reproducibility of findings. In the bottom blot assessing relative
with K14E6WT, P = 0.058, Fisher's exact test.
amounts of E6 protein in K14E6WT and K14E6D146-151 mice, the amount of
cK14E6I128T tumors were significantly different than K14E6WT
protein loaded is indicated in each lane. Mice were treated with estrogen to
tumors, P = 0.04, Wilcoxon rank-sum test.
synchronize them in estrus, thereby eliminating variability in cervical epithelialthickness.
Cancer Res 2007; 67: (4). February 15, 2007
Multiple Properties of E6 Contribute to Cervical Cancer
Figure 2. Characterization of reproductive tumors and the proliferative index of the cervix. A, comparison of tumor sizes between K14E6WT, K14E6I128T, andK14E6D146-151 transgenic mice. B, comparison of tumor sizes between K14E7WT, K14E6WT/K14E7WT, and K14E6mutant/K14E7WT transgenic mice. C, classification ofreproductive tumors by location. Middle, a cartoon representation of the murine reproductive tract and identifies the approximate borders for determining tumor locationused in histopathologic diagnosis. Left (for K14E6WT) and right (for K14E6WT/K14E7WT), the breakdown of total percentage of tumor development by location (top )and the percentage of total area these tumors encompassed (bottom ). In K14E6I128T and K14E6D146-151, the percentage of tumors arising in the vagina was 0%and 4%, respectively (data not shown). In K14E6I128T/K14E7WT and K14E6D146-151/K14E7WT transgenic mice, the tumors that developed in the vagina were 45% and38%, respectively (data not shown). D, quantification of epithelial hyperplasia in the cervix. The average percentage of basal and suprabasal BrdUrd-positive cellswas obtained from eight (!40) microscopic fields per mouse. An average of at least three mice per genotype were used to calculate the percentage.
K14E6WT mice. Thus, the ability of E6 to interact with a-helix
both the K14E6I128T/K14E7WT and the K14E6D146-151/K14E7WT
partners contributes to both tumor incidence and tumor size.
transgenic mice developed significantly smaller tumors in contrast
The interactions of E6 with both A-helix and PDZ partners
to the K14E6WT/K14E7WT mice (P = 0.005 and 0.014, respectively;
contribute to tumor size and tumor multiplicity in the
Table 2). In addition, the total area of tumor invasion of both the
reproductive tract in the presence of E7. To understand the
K14E6I128T/K14E7WT and the K14E6D146-151/K14E7WT transgenic
role of a-helix and PDZ partners of HPV16 E6 in cervical cancer
mice were significantly reduced relative to K14E6WT/K14E7WT
when E6 is expressed together with HPV16 E7, the same
tumors (P = 0.003 and 0.03, respectively; Table 2). Average tumor
K14E6mutant lines were crossed onto the K14E7WT background
size was also reduced in K14E6I128T/K14E7WT mice (data not
and treated with estrogen for 9 months. All K14E7WT mice
shown). Differences were also observed in terms of tumor
developed cervical cancer in response to estrogen treatment (9).
frequency, with both mouse lines of K14E6mutant/K14E7WT having
Therefore, it was not surprising that nearly all treated K14E6WT/
reduced number of tumors compared with K14E6WT/K14E7WT
K14E7WT and K14E6mutant/K14E7WT mice also developed cervical
mice (Table 2). This reduction was highly significant (P = 0.003)
cancer (Table 2). Differences were observed, however, in terms of
for the K14E6D146-151/K14E7WT mice, but less so for K14E6I128T/
the size of tumors. Comparing the largest tumor from each mouse,
K14E7WT mice (P = 0.187).
Cancer Res 2007; 67: (4). February 15, 2007
E6 tumors develop primarily in the cervix and the cervi-
stroma, thereby prohibiting the ability to dissect tumors for
covaginal junction. In prior studies, estrogen-treated K14E7WT
Western analyses. Hyperplastic reproductive epithelium from
transgenic mice efficiently developed tumors in the vagina as well
estrogen-treated nontransgenic mice expressed MCM7 only in
as in the cervix (9). In contrast, nearly all tumors arising in
the basal layer. In contrast, highly dysplastic reproductive
K14E6WT and K14E6mutant lines developed in the cervix or at the
epithelium from K14E6WT/K14E7WT and K14E7WT mice was
junction of the cervix and the vagina. Only 2 of 62 (6%) tumors
strongly positive for MCM7 throughout the full thickness of the
observed in the three E6 transgenic lines developed in the vagina
epithelium, similar to data from our previous studies (26).
proper. In the presence of E7, the percentage of tumors arising in
Unexpectedly, approximately one third to two thirds of the
the vagina of the K14E6WT/K14E7WT and K14E6mutant/K14E7WT
epithelia from all three E6 transgenic lines stained positive for
double transgenic mice increased to 38% to 45%. Thus, E6
MCM7, beyond the basal layer of staining in the nontransgenic
predisposes animals primarily to tumors of the cervix. In contrast,
epithelium. Expression of MCM7 in the epithelia of the three E6
E7 alone or in combination with E6 induces tumors in the vagina
transgenic lines was generally uniform. No differences in staining
as well as in the cervix, with the largest tumor area predominantly
between all doubly transgenic lines were observed, presumably due
found in the cervico-vaginal junction (Fig. 2C).
to the strong induction of MCM7 in the E7-expressing tissues. In
The ability of E6 to interact with PDZ partners contributes
agreement with previous studies (26), MCM7 staining patterns
to hyperplasia in the cervical epithelium. K14E6WT transgenic
predominantly correlated positively with lesion grade. These
mice display epidermal hyperplasia characterized by an induction
results indicate that E6 can induce Mcm7, an E2F-responsive gene.
of DNA synthesis within that suprabasal compartment (5). A
In tumors, MCM7 expression had a less consistent pattern
similar finding was observed in the cervical epithelium (Fig. 2D),
compared with the epithelium. Expression of MCM7 in tumors was
with significant increases in both basal and suprabasal DNA
variable, not correlating with genotype, tumor size, or location.
synthesis (P = 0.02) in K14E6WT transgenic mice (11.6% and 2.9%,
Furthermore, levels varied between tumors arising within the same
respectively) compared with nontransgenic mice (5.6% and 0.9%,
mouse. The sole tumor arising in the nontransgenic mouse had low
respectively). Suprabasal DNA synthesis in the K14E6D146-151
MCM7 expression (Fig. 3; Table 3). Nonetheless, all tumors from
transgenic mice was reduced compared with K14E6WT transgenic
transgenic mice had some level of MCM7 expression. No tumor
mice (1.64% versus 2.87%; P = 0.06) and not significantly different
was MCM7 negative. Aside from K14E7WT tumors, which were
from that observed in nontransgenic mice (P = 0.14). No difference
robust for MCM7 staining, all tumors from other genotypes had
in suprabasal DNA synthesis was observed between K14E6I128T and
low to high MCM7 expression.
K14E6WT transgenic mice (P = 0.26). Thus, in the cervical
Cyclin E is another E2F-responsive gene that is also used as a
epithelium, the E6 oncogene is able to increase DNA synthesis in
biomarker for dysplastic lesions and cervical cancer (27). We
both basal and suprabasal layers of the cervical epithelium,
evaluated cyclin E expression using immunohistochemistry in the
resulting in hyperplasia. This is comparable with results in the
epithelia and in tumors for all genotypes. In the absence of E7,
skin, where the ability of E6 to increase suprabasal DNA synthesis
cyclin E staining was more nuclear and less cytoplasmic. In
was mediated at least partially through interactions with PDZ
nontransgenic mice, cyclin E expression was restricted mostly to
partners (10).
the basal and parabasal layers of cervical epithelia. In both
Mcm7 and cyclin E are up-regulated in E6 epithelia and
K14E6WT and K14E6mutant cervical epithelium, cyclin E expression
tumors of the reproductive tract in the absence of E7. Mcm7 is
positively correlated with the level of dysplasia (data not shown).
an E2F-responsive gene and robust biomarker expressed in high-
Unlike MCM7, cyclin E expression was not uniform, such that not
grade CINs and cervical cancer in humans as well as in K14E6WT/
every cell was positive. The sole nontransgenic tumor had low
K14E7WT and K14E7WT mice (26). We evaluated MCM7 expression
cyclin E expression. Tumors from the singly E6 transgenic lines had
in the epithelia and in tumors of the reproductive tract arising in
variable cyclin E expression similar to that observed for MCM7.
nontransgenic, K14E7WT and the three E6 transgenic lines with or
Tumors had low to high cyclin E expression with no correlation
without E7 (Fig. 3). Analyses of biomarker expression in both the
between staining level, tumor size, or location.
epithelium and the tumors were limited to immunohistochemistry.
In the presence of E7, cyclin E diffusely stained both nuclei and
Reproductive tract tumors in HPV transgenic mice were often too
cytoplasm (Fig. 3; Table 3). K14E7WT reproductive epithelium had
small and sessile. The tumors tended to grow inwardly into the
at least 50% of cells staining positive for cyclin E. In general,
Table 2. Comparison of tumors from the reproductive tract of nontransgenic, K14E7WT, K14E6WT/K14E7WT, andK14E6mutant/K14E7WT transgenic mice
Total area of tumor
K14E6WT/K14E7WT (n = 6)
K14E6I128T/K14E7WT (n = 24)
K14E6D146-151/K14E7WT (n = 21)
*Compared with K14E6WT/K14E7, P < 0.05, Wilcoxon rank-sum test.
Cancer Res 2007; 67: (4). February 15, 2007
Multiple Properties of E6 Contribute to Cervical Cancer
Figure 3. Evaluation of E2F-responsivegene expression in the estrogen-treatedepithelium and tumors from thereproductive tract. Columns 1 and 2,MCM7 staining (brown staining nuclei),which is up-regulated in singly or doubletransgenic reproductive epithelia andtumors, whereas MCM7 expression isrestricted to the basal and parabasal layersof nontransgenic epithelium. Columns 3and 4, cyclin E staining (brown nuclei).
Cyclin E expression, similar to MCM7expression, is also up-regulated in bothsingle and double transgenic mice.
Magnification, !40.
E7-positive tumors had medium to high cyclin E expression. Cyclin
was variable, with a range from nil to sporadic highly positive
E expression in epithelia and tumors of doubly transgenic mice was
(Fig. 4). Tumors from K14E6mutant/K14E7WT transgenic mice (data
generally high, with at least 60% of cells staining positive.
not shown) had similar levels of p53 expression relative to
Reproductive tract tumors are more likely to express p53
K14E6WT/K14E7WT tumors but were generally less intense. We
and in greater intensities in the presence of E7. p53 is generally
also noted that vaginal tumors had reduced expression of p53
undetectable in normal tissue unless induced in response to DNA-
relative to tumors from the cervix or the cervicovaginal junction.
damaging agents. The reproductive tract is a p53-responsive tissue,
p16 expression is inversely correlated with retinoblastoma
in which a DNA damage response can be mounted in response to
expression in reproductive tumors from HPV transgenic mice.
ionizing radiation if WT p53 is intact. Dominant-acting, missense
p16 is up-regulated in several cervical cancer cell lines as well as in
mutations in p53, causally associated with tumorigenesis, often
human cervical samples (23). This cyclin kinase inhibitor is a
lead to the stabilization and accumulation of p53, providing a
marker for high-risk HPV infection in human dysplastic lesions and
useful indicator of p53 status and the disease state in most tumor
cancers of the reproductive tract as well as cancers of the head and
types. In HPV-associated cancers, p53 is thought to be inactivated
neck (31, 32). We evaluated p16 status in the tumors from our
through E6-induced, ubiquitin-mediated protein degradation.
However, p53 mutations have been detected in both premalignantlesions and human cervical tumors at low frequencies. We did p53
Table 3. Summary of biomarker expression in tumors
immunohistochemistry to monitor levels of p53 protein in the
from the reproductive tract of estrogen-treated mice
reproductive tumors of our various HPV transgenic mice (Fig. 4;Table 3). The sole nontransgenic tumor was p53 negative. Tumors
from K14E7WT transgenic mice were p53 positive and had low tomedium expression, whereas tumors from K14E6WT transgenic
mice were almost completely p53 negative. These observations
were consistent with prior studies showing the destabilization of
p53 by E6- and E7-induced accumulation of p53 (28–30).
K14E6I128T-expressing epithelium had elevated p53 expression
compared with K14E6WT mice, consistent with the reduced ability
of K14E6I128T to degrade p53. Tumors from K14E6I128T mice,
however, had clearly less intense levels of p53 expression thantumors from K14E7WT mice. In agreement with our predictions, theK14E6D146-151 transgenic tumors were p53 negative. Expression of
NOTE: ", negative; F, <5%; +, 5% to 20%; ++, 20% to 50%; +++, >50%.
p53 in the tumors arising in K14E6WT/K14E7WT transgenic mice
Cancer Res 2007; 67: (4). February 15, 2007
estrogen-treated mice. Expression of p16 was uniformly diffuse
(Fig. 4; Table 3). The sole nontransgenic tumor had low expres-
with both nuclear and cytoplasmic expression in all mice. There
sion levels of pRb. In agreement with the known ability of E7 to
was a general cytoplasmic expression pattern, with nuclear
induce the degradation of pRb, E7-expressing tumors had little
accumulation localized predominantly to the bottom one third to
to no detectable pRb regardless of E6 mutational status (Fig. 4;
one half of the epithelium (data not shown). The sole non-
Table 3; data not shown). In contrast, tumors from K14E6WT and
transgenic tumor had low levels of p16 expression. Tumors from
K14E6D146-151 transgenic mice expressed high levels of pRb.
both K14E6WT/K14E7WT and K14E7WT mice displayed high levels of
K14E6I128T tumors, however, displayed low levels of pRb relative
p16 (Fig. 4; Table 3), similar to the patterns observed in human
to K14E6WT and K14E6D146-151 tumors. Hence, HPV E6 is able to
cervical samples. Presumably, due to the ability of E7 to induce p16
alter pRb and p16 levels in tumors in a manner distinguishable
strongly (33), differences in expression between K14E6mutant/
from HPV E7 and is dependent on its interaction with a-helix
K14E7WT tumors were not observed (data not shown). Conversely,
partners, as the expression pattern of p16 and pRb no longer
expression of p16 in tumors from the K14E6WT and K14E6D146-151
resembles WT E6, but more like E7-expressing tumors.
transgenic mice was either low or negative. This decrease in p16expression was clearly more pronounced in the tumors comparedwith the neighboring epithelium, which was variable (data not
shown). In contrast, tumors from K14E6I128T transgenic mice
In this study, we dissected the contributions of HPV E6 in both
displayed a strong increase in the expression of p16 relative to
the presence and the absence of HPV E7 in cervical carcinogenesis
K14E6WT and K14E6D146-151 tumors. The intensity of p16 expression
by focusing on specific properties of E6 and extending the estrogen
in K14E6I128T tumors was generally less robust relative to tumors
treatment period. E6, in the absence of E7, induces primarily
containing the E7 oncogene.
cervical tumors in the reproductive tract. The abilities of E6 to
Because expression of p16 was reduced in K14E6 reproductive
interact with both a-helix and PDZ partners contribute to this role
tumors and given that pRb contributes to the regulation of p16, we
in cervical carcinogenesis. Furthermore, E6 induces a pattern of
measured the expression levels of pRb via immunohistochemistry
cellular gene expression that is overlapping yet distinct from that
Figure 4. p53, p16, and pRb status in thereproductive tract. Images from sectionsstained with antibodies to p53, p16, orpRb (brown ) and counterstained withhematoxylin (blue ). Column 1, p53expression in the cervical epithelium fromvarious genotypes. The nontransgenic(NTG ; top ) sample is from a mouseexposed to 5 Gy of ionizing radiation andis used as a positive control to showp53-positive staining, which is primarilyobserved in the basal and parabasal strata.
The cervical epithelium from unirradiatednontransgenic mice (data not shown) isp53 negative. All other panels are fromunirradiated mice. Columns 2 to 4, thestatus of p53, p16, and retinoblastoma(pRB ) expression in reproductive tumorsfrom various genotypes. Tumorsexpressing the E7 oncogene generallydisplayed variable positivity for p53. Shownin the panel from the E6/E7 tumor is anarea of high sporadic p53 positivity. p16was up-regulated in tumors expressing E7or K14E6I128T. Retinoblastoma expressionwas inversely correlated to p16 in tumorsexpressing either HPV E6 or E7.
Magnification, !40.
Cancer Res 2007; 67: (4). February 15, 2007
Multiple Properties of E6 Contribute to Cervical Cancer
tumors of K14E6WT mice. Whether this alternative dysregulation of
Table 4. Summary of histopathologic diagnosis in
the p16/pRb pathway contributes to the tumorigenesis observed in
K14E6WT and K14E6mutant transgenic mice
the K14E6I128T transgenic mice is unclear. Regardless, this findingsupports the hypothesis that residual oncogenic activity in the
CIN I CIN II CIN III/CIS Cancer
K14E6I128T mice reflects a distinct activity of E6 and not a partial
retention in the binding capacity to a-helix partners. The absence
K14E6I128T (n = 36)
of a reduction in the tumorigenic phenotype of K14E6D146-151
K14E6D146-151 (n = 28)
compared with K14E6WT mice indicates that PDZ domain partnersare not relevant in the context of these experiments or that theircontribution is modest.
Abbreviations: CIS, carcinoma in situ; NH, normal hyperplasia.
The contribution of E6 to cervical carcinogenesis in the
presence of E7 is dependent on interactions with both A-helixand PDZ partners. The above experiments were all carried out inthe absence of E7. Similar studies in the presence of E7 (Table 2)
induced by E7. Specifically, E6 leads to a dysregulation of the p16/
revealed that the interaction of E6 with a-helix partners is
pRb pathway in a manner different from that of E7 yet led to a
important for cervical carcinogenesis. The most obvious difference
similar though less robust induction of E2F-responsive genes.
in the tumorigenic phenotypes between K14E6 I128T/K14E7WT and
HPV16 E6 has a weaker oncogenic potential than HPV16 E7
K14E6WT/K14E7WT mice was tumor size. In contrast to observa-
in the reproductive tract. In our prior studies, K14E6WT
tions in the absence of E7, the interactions of E6 with PDZ partners
transgenic mice did not develop cervical cancer or even high-
also contributed to cervical carcinogenesis in the presence of E7.
grade dysplasias after 6 months of estrogen treatment. K14E7WT
Specifically, we observed a reduction in tumor size in the
transgenic mice on the hand developed multiple high-grade
K14E6D146-151/K14E7WT mice compared with K14E6WT/K14E7WT
dysplastic lesions and tumors throughout the entire reproductive
mice. Tumors arising in the K14E6mutant/K14E7WT mice were not
tract. In this study, we extended the estrogen treatment period to
significantly different in size from those arising in the K14E7WT
9 months. A large fraction (41%) of K14E6WT transgenic mice
singly transgenic mice. Tumor multiplicity also was reduced in
developed cancer. The majority of the remaining K14E6 mice
both K14E6mutant/K14E7WT lines relative to K14E6WT/K14E7WT
developed at least one high-grade dysplastic lesion (Table 4). This
mice. This reduction was only statistically significant for the
represented a significant increase in tumorigenesis compared with
K14E6D146-151/K14E7WT transgenic mice. Thus, in the presence of
nontransgenic mice (6.7%) yet significantly less than that observed
E7, E6 contributes to cervical carcinogenesis through at least two
in K14E7WT mice (100% tumor incidence). Likewise, tumor
distinct mechanisms. This finding is consistent with what we have
multiplicity was significantly reduced in K14E6WT mice compared
observed previously in the skin, where the ability of E6 to bind both
with the K14E7WT mice (1.67 versus 6.67). Furthermore, the
a-helix and PDZ domain partners contributed to carcinogenesis
K14E6WT transgenic mice did not develop the extensive dysplastic
(10, 17, 19).
pathology that occurred throughout the entire reproductive
It is unclear which PDZ partner(s) of E6 contributes to both
epithelial lining of K14E7WT mice. Thus, HPV16 E6 has a
tumor size and tumor multiplicity. The E6 oncogene interacts with
demonstrable yet clearly weaker oncogenic activity than HPV16
numerous cellular partners, which contain PDZ motifs, such as Dlg,
E7 in the reproductive tract. In contrast, E6 is the more potent
Scribble, and Magis (34–37). DLG and/or Scribble are attractive
oncogene in the skin, contributing to both the promotion and the
candidates given that both are tumor suppressors in Drosophila.
progression stages of skin carcinogenesis and induces primarily
Mutations in either of these genes in Drosophila result in the
malignant tumors (8). Therefore, the relative potency of the HPV16
development of epithelial hyperplasia, loss of cell-cell contacts
E6 and E7 oncogenes differs depending on the tissue evaluated.
(38, 39), and tumorigenesis of the imaginal discs and brain lobes
The ability of E6 to bind to A-helix partners contributes to
(40). In the human cervix, both hDlg and hScrib are gradually
cervical carcinogenesis. K14E6I128T transgenic mice had a
altered in cellular localization and expression is lost as low-grade
reduction in the incidence and size of reproductive tract tumors
lesions progress to invasive cervical carcinomas (41–43). Reduc-
compared with K14E6WT transgenic mice. Given the reduced
tions in hDlg and hScrib are also seen in human cervical cancer cell
tumorigenic phenotype of the K14E6I128T transgenic mice, we
lines (19). Because Dlg and Scribble are both expressed in the
hypothesize that the inactivation of p53 by E6 contributes to
septate junction (44) and seem to have similar functions, both
cervical carcinogenesis. Consistent with this hypothesis, slightly
genes may contribute to the oncogenic potential of E6 in the
elevated levels of p53 protein were seen in tumors arising in
reproductive tract. Until analysis of targeted individual PDZ
K14E6I128T mice compared with K14E6WT mice.
deletion mutants can be done, the exact E6-PDZ interaction(s)
Whereas reduced in their incidence of tumors compared with
responsible for the oncogenic potential of E6 remains unclear.
K14E6WT mice, K14E6I128T transgenic mice retained a significant
The E6 oncogene induces the E2F-responsive genes, MCM7
increase in their tumorigenic phenotype compared with non-
and cyclin E, in reproductive epithelia and tumors in the
transgenic mice (Table 1). This increase was evident in tumor
absence of E7. All E6 transgenic lines expressed the E2F-
multiplicity (1.14 versus 0.07) and average tumor size (1.39 mm2
responsive genes, MCM7 and cyclin E, in both the epithelium
versus 0.029 mm2). Such residual oncogenic activity in K14E6I128T
and the tumors of the reproductive tract. Expression of these genes
transgenic mice could reflect the ability of the I128T mutant
was above the levels seen in nontransgenic mice. These results in
protein to bind a-helix partners, albeit at 1% to 5% the level of WT
the E6 mice were somewhat unexpected given that the E7
E6 protein, or it may reflect an activity of E6 distinct from its ability
oncogene was absent in these mice and therefore not available
to bind a-helix partners. Tumors arising in K14E6I128T mice also
to induce the expression of E2F-responsive genes through pRb
displayed a distinct pattern of expression of p16 and pRb relative to
inactivation. Hence, the E6 oncogene must be activating the
Cancer Res 2007; 67: (4). February 15, 2007
transcription of these E2F-responsive genes by a mechanism
How E6 induces levels of CDK4/6 is unknown. Thus, it remains
different than E7. In the epithelium of the K14E6WT mice, this up-
unclear whether these two hypotheses reflect the same or distinct
regulation of E2F-responsive genes correlated with high levels of
mechanisms. Regardless, a role of p53 inactivation in mediating
p16 and low levels of pRb, as seen in the epithelium and tumors in
the dysregulation of p16/pRb pathway by E6 is supported by the
the K14E7WT mice. However, there is an inverted pattern of
reversed pattern of p16 expression in K14E6I128T tumors, which
expression of p16 and pRb in the K14E6WT tumors (Fig. 4; Table 3).
encode a mutant E6 protein defective for inactivating p53 compared
Specifically, levels of p16 were low and levels of pRb were high in
with K14E6WT tumors (Fig. 4; Table 3).
the reproductive tumors of K14E6WT mice. This result indicates
In summary, we report the first in vivo study dissecting the
that the alteration of the cell cycle during progression to
mechanism of E6 action in cervical carcinogenesis. The E6 inter-
malignancy in K14E6WT mice differs from that observed in
actions with two groups of cellular partners contributed to cervical
K14E7WT mice. Interestingly, the pattern seen in the tumors of
carcinogenesis. Additionally, our study revealed that the ability
K14E6WT mice is consistent with the low expression levels of p16
of E6 to induce E2F-responsive genes is likely through the dys-
and high levels of hyperphosphorylated pRb observed in fibroblast
regulation of the p16/pRb pathway by mechanisms distinct from
and epithelial cell lines immortalized with the HPV E6 oncogene
(33, 45–47). Thus, E6-dependent immortalization in vitro and E6-dependent tumorigenesis in vivo arise through means that lead to asimilar dysregulation of the p16/pRb pathway opposite of that
observed in E7-dependent tumorigenesis (this study) or in human
Received 9/8/2006; revised 10/30/2006; accepted 12/4/2006.
cervical cancers (48). The inactivation of p53 by E6 and consequent
The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance
inhibition of p53-induced expression of the cyclin-dependent kinase
with 18 U.S.C. Section 1734 solely to indicate this fact.
(CDK) inhibitor p21 might lead to higher CDK activity and thereby
We thank Dr. E. Weiss (University Louis Pasteur, Strasbourg, France) for generating
and providing the anti-E6 monoclonal antibody 6F4, Drs. Drinkwater and Sugden for
increased hyperphosphorylated pRb. Alternatively, E6 could induce
the critical reading of the manuscript, and members of the Lambert laboratory for
phosphorylation of pRb by up-regulating the levels of CDK4/6 (49).
13. Huibregtse JM, Scheffner M, Beaudenon S, Howley
25. Gulliver GA, Herber RL, Liem A, Lambert PF. Both
PM. A family of proteins structurally and functionally
conserved region 1 (CR1) and CR2 of the human
1. Sankaranarayanan R, Ferlay J. Worldwide burden of
related to the E6-AP ubiquitin-protein ligase. Proc Natl
papillomavirus type 16 E7 oncogene are required for
gynaecological cancer: the size of the problem. Best
Acad Sci U S A 1995;92:5249.
induction of epidermal hyperplasia and tumor forma-
Pract Res Clin Obstet Gynaecol 2006;20:207–25.
14. Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP
tion in transgenic mice. J Virol 1997;71:5905–14.
2. Walboomers JM, Jacobs MV, Manos MM, et al. Human
mutations cause Angelman syndrome. Nat Genet 1997;
26. Brake T, Connor JP, Petereit DG, Lambert PF.
papillomavirus is a necessary cause of invasive cervical
Comparative analysis of cervical cancer in women and
cancer worldwide. J Pathol 1999;189:12–9.
15. Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A,
in a human papillomavirus-transgenic mouse model:
3. Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV.
Ledbetter DH, Beaudet AL. Imprinting analysis of three
identification of minichromosome maintenance protein
The causal relation between human papillomavirus and
genes in the Prader-Willi/Angelman region: SNRPN, E6-
7 as an informative biomarker for human cervical
cervical cancer. J Clin Pathol 2002;55:244–65.
associated protein, and PAR-2 (D15S225E). Hum Mol
cancer. Cancer Res 2003;63:8173–80.
4. Fehrmann F, Laimins LA. Human papillomaviruses:
27. Keating JT, Cviko A, Riethdorf S, et al. Ki-67, cyclin E,
targeting differentiating epithelial cells for malignant
16. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM.
and p16INK4 are complimentary surrogate biomarkers
transformation. Oncogene 2003;22:5201–7.
The HPV-16 E6 and E6-AP complex functions as a
for human papilloma virus-related cervical neoplasia.
5. Song S, Pitot HC, Lambert PF. The human
ubiquitin-protein ligase in the ubiquitination of p53. Cell
Am J Surg Pathol 2001;25:884–91.
papillomavirus type 16 E6 gene alone is sufficient to
28. Scheffner M, Werness BA, Huibregtse JM, Levine AJ,
induce carcinomas in transgenic animals. J Virol 1999;
17. Nguyen M, Song S, Liem A, Androphy E, Liu Y,
Howley PM. The E6 oncoprotein encoded by human
Lambert PF. A mutant of human papillomavirus type 16
papillomavirus types 16 and 18 promotes the degrada-
6. Herber R, Liem A, Pitot H, Lambert PF. Squamous
E6 deficient in binding a-helix partners displays reduced
tion of p53. Cell 1990;63:1129–36.
epithelial hyperplasia and carcinoma in mice transgenic
oncogenic potential in vivo . J Virol 2002;76:13039–48.
29. Jones DL, Thompson DA, Munger K. Destabilization
for the human papillomavirus type 16 E7 oncogene.
18. Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway
of the RB tumor suppressor protein and stabilization of
J Virol 1996;70:1873–81.
DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and
p53 contribute to HPV type 16 E7-induced apoptosis.
7. Riley RR, Duensing S, Brake T, Munger K, Lambert PF,
telomerase activity are required to immortalize human
Arbeit JM. Dissection of human papillomavirus E6 and
epithelial cells. Nature 1998;396:84–8.
30. Eichten A, Westfall M, Pietenpol JA, Munger K.
E7 function in transgenic mouse models of cervical
19. Simonson SJ, Difilippantonio MJ, Lambert PF. Two
Stabilization and functional impairment of the tumor
carcinogenesis. Cancer Res 2003;63:4862–71.
distinct activities contribute to human papillomavirus
suppressor p53 by the human papillomavirus type 16 E7
8. Song S, Liem A, Miller JA, Lambert PF. Human
16 E6's oncogenic potential. Cancer Res 2005;65:8266–73.
oncoprotein. Virology 2002;295:74–85.
papillomavirus types 16 E6 and E7 contribute differently
20. Tachibana KE, Gonzalez MA, Coleman N. Cell-cycle-
31. Sano T, Oyama T, Kashiwabara K, Fukuda T,
to carcinogenesis. Virology 2000;267:141–50.
dependent regulation of DNA replication and its
Nakajima T. Expression status of p16 protein is
9. Brake T, Lambert PF. Estrogen contributes to the
relevance to cancer pathology. J Pathol 2005;205:123–9.
associated with human papillomavirus oncogenic po-
onset, persistence, and malignant progression of cervi-
21. Kim Y, Choi EK, Cho NH, et al. Expression of cyclin E
tential in cervical and genital lesions. Am J Pathol 1998;
cal cancer in a human papillomavirus-transgenic mouse
and p27KIP1 in cervical carcinoma. Cancer Lett 2000;
model. Proc Natl Acad Sci U S A 2005;102:2490–5.
32. Hafkamp HC, Speel EJ, Haesevoets A, et al. A subset
10. Nguyen ML, Nguyen MM, Lee D, Griep AE,
22. Erlandsson F, Martinsson-Ahlzen HS, Wallin KL,
of head and neck squamous cell carcinomas exhibits
Lambert PF. The PDZ ligand domain of the human
Hellstrom AC, Andersson S, Zetterberg A. Parallel cyclin
integration of HPV 16/18 DNA and overexpression of
papillomavirus type 16 E6 protein is required for E6's
E and cyclin A expression in neoplastic lesions of the
p16INK4A and p53 in the absence of mutations in p53
induction of epithelial hyperplasia in vivo . J Virol
uterine cervix. Br J Cancer 2006;94:1045–50.
exons 5-8. Int J Cancer 2003;107:394–400.
23. Klaes R, Friedrich T, Spitkovsky D, et al. Over-
33. Khleif SN, DeGregori J, Yee CL, et al. Inhibition of
11. Nguyen MM, Nguyen ML, Caruana G, Bernstein A,
expression of p16(INK4A) as a specific marker for
cyclin D-CDK4/CDK6 activity is associated with an E2F-
Lambert PF, Griep AE. Requirement of PDZ-containing
dysplastic and neoplastic epithelial cells of the cervix
mediated induction of cyclin kinase inhibitor activity.
proteins for cell cycle regulation and differentiation in the
uteri. Int J Cancer 2001;92:276–84.
Proc Natl Acad Sci U S A 1996;93:4350–4.
mouse lens epithelium. Mol Cell Biol 2003;23:8970–81.
24. Giovane C, Trav G, Briones A, Lutz Y, Wasylyk B,
34. Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T,
12. Liu Y, Chen JJ, Gao Q, et al. Multiple functions of
Weiss E. Targeting of the N-terminal domain of the
Ishibashi M. Binding of high-risk human papillomavirus
human papillomavirus type 16 E6 contribute to the
human papillomavirus type 16 E6 oncoprotein with
E6 oncoproteins to the human homologue of the
immortalization of mammary epithelial cells. J Virol
monomeric ScFvs blocks the E6-mediated degradation
Drosophila discs large tumor suppressor protein. Proc
of cellular p53. J Mol Recognit 1999;12:141–52.
Natl Acad Sci U S A 1997;94:11612–6.
Cancer Res 2007; 67: (4). February 15, 2007
Multiple Properties of E6 Contribute to Cervical Cancer
35. Gardiol D, Kuhne C, Glaunsinger B, Lee SS, Javier R,
and Lgl in cell polarity, cell proliferation, and cancer.
cence and loss of p16 at immortalization in human
Banks L. Oncogenic human papillomavirus E6 proteins
papillomavirus 16 E6, but not E7, transformed human
target the discs large tumour suppressor for protea-
41. Lin HT, Steller MA, Aish L, Hanada T, Chishti AH.
uroepithelial cells. Cancer Res 1996;56:2886–90.
some-mediated degradation. Oncogene 1999;18:5487–96.
Differential expression of human Dlg in cervical intra-
46. Yamamoto A, Kumakura S, Uchida M, Barrett JC,
36. Nakagawa S, Huibregtse JM. Human scribble (Vartul)
epithelial neoplasias. Gynecol Oncol 2004;93:422–8.
Tsutsui T. Immortalization of normal human embryonic
is targeted for ubiquitin-mediated degradation by the
42. Cavatorta AL, Fumero G, Chouhy D, et al. Differential
fibroblasts by introduction of either the human
high-risk papillomavirus E6 proteins and the E6AP
expression of the human homologue of drosophila discs
papillomavirus type 16 E6 or E7 gene alone. Int J Cancer
ubiquitin- protein ligase. Mol Cell Biol 2000;20:8244–53.
large oncosuppressor in histologic samples from human
37. Thomas M, Laura R, Hepner K, et al. Oncogenic
papillomavirus-associated lesions as a marker for
47. Tsutsui T, Kumakura S, Yamamoto A, et al. Association
human papillomavirus E6 proteins target the MAGI-2
progression to malignancy. Int J Cancer 2004;111:373–80.
of p16(INK4a) and pRb inactivation with immortalization
and MAGI-3 proteins for degradation. Oncogene 2002;
43. Nakagawa S, Yano T, Nakagawa K, et al. Analysis of
of human cells. Carcinogenesis 2002;23:2111–7.
the expression and localisation of a LAP protein, human
48. Nam EJ, Kim JW, Kim SW, et al. The expressions of
38. Bilder D, Li M, Perrimon N. Cooperative regulation of
scribble, in the normal and neoplastic epithelium of
the Rb pathway in cervical intraepithelial neoplasia;
cell polarity and growth by Drosophila tumor suppres-
uterine cervix. Br J Cancer 2004;90:194–9.
predictive and prognostic significance. Gynecol Oncol.
sors. Science 2000;289:113–6.
44. Tepass U, Tanentzapf G, Ward R, Fehon R. Epithelial
Epub 2006 Oct 13.
39. Bilder D, Perrimon N. Localization of apical epithelial
cell polarity and cell junctions in Drosophila . Annu Rev
49. Malanchi I, Accardi R, Diehl F, et al. Human
determinants by the basolateral PDZ protein Scribble.
papillomavirus type 16 E6 promotes retinoblastoma
45. Reznikoff CA, Yeager TR, Belair CD, Savelieva E,
protein phosphorylation and cell cycle progression.
40. Humbert P, Russell S, Richardson H. Dlg, Scribble,
Puthenveettil JA, Stadler WM. Elevated p16 at senes-
J Virol 2004;78:13769–78.
Cancer Res 2007; 67: (4). February 15, 2007
Source: http://beats.datasynth.info/wp-content/uploads/2016/03/Shai_etal_2007.pdf
Drug Facts Labels Allegra® Allergy 180-mg Tablets Drug FactsActive ingredient (in each tablet) Fexofenadine HCl 180 mg .Antihistamine temporarily relieves these symptoms due to hay fever or other upper respiratory allergies:• runny nose • itchy, watery eyes • itching of the nose or throat Do not use if you have ever had an allergic reaction to this product or any of its ingredients.Ask a doctor before use if you have kidney disease. Your doctor should determine if you
Account Number: 1234 5678 9012 3456 April 16, 2012 to May 16, 2012 Account InformationDays in Bil ing Cycle Statement Closing Date Available Credit as of 05/16/12 Purchases & Other Charges Payment Due Date Fees Charged Amount Over Limit Due Total Minimum Amount Due Late Payment Warning: If we do not receive your minimum payment and past due amount by the date listed above, you may have to pay a late fee of up to


