
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Chimiotheque-nationale.cn.cnrs.fr



Biochem. J. (2012) 443, 549–559 (Printed in Great Britain)
First identification of small-molecule inhibitors of Pontin by combining
virtual screening and enzymatic assay
Judith ELKAIM*, Michel CASTROVIEJO†, Driss BENNANI*, Said TAOUJI‡, Nathalie ALLAIN‡, Michel LAGUERRE*,Jean ROSENBAUM‡, Jean and Patrick LESTIENNE*Molecular Modeling Group, IECB-CNRS-Universit´e de Bordeaux, UMR 5248, 2 rue R. Escarpit, F-33607 Pessac, France, †Platform Protein Expression and Purification, CNRS, UMR5234, 146 rue L. Saignat, F-33076 Bordeaux Cedex, France, and ‡Physiopathologie du Cancer du Foie, INSERM U1053-Universit´e de Bordeaux, 146 rue L. Saignat, F-33076 BordeauxCedex, France
The human protein Pontin, which belongs to the AAA +
and sensitive colorimetric assay was set up to measure the dis-
(ATPases associated with various cellular activities) family, is
ruption of the ATPase activity of Pontin. This assay allowed the
overexpressed in several cancers and its silencing in vitro leads
determination of inhibition curves for more than 20 top-scoring
to tumour cell growth arrest and apoptosis, making it a good
compounds, resulting in the identification of four ligands present-
target for cancer therapy. In particular, high levels of expression
ing an inhibition constant in the micromolar concentration range.
were found in hepatic tumours for which the therapeutic arsenal
Three of them inhibited tumour cell proliferation. The association
is rather limited. The three-dimensional structure of Pontin has
of virtual screening and experimental assay thus proved successful
been resolved previously, revealing a hexameric assembly with
for the discovery of the first small-molecule inhibitors of Pontin.
one ADP molecule co-crystallized in each subunit. Using Vina,DrugScore and Xscore, structure-based virtual screening of 2200commercial molecules was conducted into the ATP-binding site
formed by a dimer of Pontin in order to prioritize the best
small-molecule inhibitor, TATA-box-interacting protein 49
candidates. Complementary to the in silico screening, a versatile
(TIP49), Vina, virtual screening.
structure for the hexameric assembly was released online afterthe present paper had been submitted for review. It was therefore
Pontin, also known as TIP (TATA-box-binding protein) 49
not taken into account in our simulations.) In fact, the proposed
or RUVBL (RuvB-like) 1, and its homologue Reptin (TIP48,
structure presents a heterohexamer composed of alternating
RUVBL2), with which it shares 40 % sequence identity and
subunits of Pontin and Reptin which will probably have a great
65 % homology belong to the AAA + (ATPase associated
significance in the mechanistic comprehension of this complex.
with various cellular activities) family and display homo-
Most publications agree that human Pontin and Reptin are
logies with the bacterial RuvB helicase The structure of
indeed endowed with an ATPase activity either alone, or in
Pontin shows characteristic ATPase Walker A and B domains;
heteromeric complexes Pontin and Reptin belong to several
the Walker A motif plays a role in nucleotide binding and in
multi-protein complexes in the nucleus where they are
metal-ion co-ordination, whereas the Walker B domain contains
thought to participate in chromatin remodelling double-
residues involved in metal-ion co-ordination and ATP hydrolysis.
strand break DNA repair and regulation of transcription
Additional domains such as the sensors 1 and 2, and the arginine
They notably interact with the oncogenic transcription factors β-
finger (Arg357) interact with the γ -phosphate and play an important
catenin and c-Myc and modulate their activities They are
role in intersubunit communication and interactions The
also involved in the biogenesis and assembly of ribonucleoprotein
arrangement in hexameric complexes, shared with other AAA +
complexes such as snoRNPs (small nucleolar ribonucleoproteins)
family members was deduced from X-ray diffraction analysis
and from electron microscopy experiments either for Pontin
We have reported previously that both Pontin and Reptin
alone or in complex with Reptin in yeast and human The
were overexpressed in human hepatocellular carcinoma.
three-dimensional reconstitution of electron microscopy images
Overexpression of these proteins was also found in a number
revealed a dodecameric edifice with a central cavity forming a
of other human cancers In vitro silencing of either Pontin or
tunnel compatible with the diameter of potential polynucleotides.
Reptin led to tumour cell growth reduction Furthermore,
As confirmed by the crystallographic structure of Pontin alone
in vivo silencing of Reptin in xenografted tumours dramatically
published by Matias et al. in 2006 this complex was supposed
reduced tumour progression These features indicate that
to be composed of a homohexamer of Pontin superimposed on
Pontin and Reptin could be good candidates for cancer therapy.
top of a homohexamer of Reptin, for which no three-dimensional
With the use of Walker A or B mutants of Pontin devoid of
structure existed. However, another structure has been recently
ATPase activity, several authors have suggested that the ATPase
described by the same team, displaying important differences with
activity of Pontin is required for growth and viability in yeast
the original one, mainly in the hexameric arrangements which are
cells The same method allowed the demonstration that
not composed of Pontin alone (This article presenting a new
the D302N Walker B mutant of Pontin did not support telomerase
Abbreviations used: AAA + , ATPase associated with various cellular activities; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-
sulfophenyl)-2H-tetrazolium; PTP1B, protein tyrosine phosphatase 1B; RUVBL, RuvB-like; TIP, TATA-box-binding protein.
1 Correspondence may be addressed to either of these authors (email [email protected] for molecular modelling aspects or
[email protected] for biochemical aspects).
� The Authors Journal compilation c
� 2012 Biochemical Society
J. Elkaim and others
biogenesis in human cells and inhibited cell transformation
CCG-3�, hairpin (44-mer), 5�-TCGCTCTTCTACTATGAACCC-
by several oncogenes such as c-Myc β-catenin or E1A
CCCTCCCCATTTTTGGGGAGGGG-3�, and double-stranded
Therefore targeting the ATPase activity of Pontin appears to
be a suitable strategy against cancer.
In the present study, we present two complementary approaches
for the discovery of inhibitors of Pontin. No report of small-
Receptor preparation for docking
molecule inhibitors for Pontin or Reptin has been made to date,
The crystal structure of Pontin bound with ADP (PDB code
since a major drawback encountered by several teams in the design
2C9O) was used for docking Except for two loops from
of an enzymatic assay for Pontin was the low activity of the
residues 142–155 and 248–276, the structure was complete.
protein, if any, when detected by the hydrolysis of radiolabelled
Those loops were far enough from the region of interest and
ATP, yielding ADP and Pi On top of that, this method
therefore were not reconstructed before calculations. The
is time-consuming owing to the use of TLC, presents difficulties
structure was visualized using Discovery Studio 2.1 (Accelrys).
because of the short half-life of 32P, and is slightly harmful
All water molecules were removed and missing hydrogen atoms
and difficult to handle in a standard laboratory. An easy-to-
were added using Charmm forcefield. A minimization of the
handle colorimetric assay that only requires a spectrophotometer
structure was conducted in the presence of bound ADP, using
was designed in order to overcome these limitations and
a Steepest Descent algorithm, 2000 steps with a 0.01 gradient,
allowed the identification of 29 inactive compounds from the
keeping the backbone of the protein fixed. The ADP-binding site
French Chimioth eque Nationale. These molecules presented no
is located at the interface between two subunits, therefore the
inhibitory effect on the ATP hydrolysis displayed by Pontin.
docking was conducted on a dimer of Pontin to take into account
In order to select potential inhibitors, we took advantage
potential interactions with the second subunit.
of the three-dimensional structure of the hexameric complexof Pontin with bound ADP, which has been resolved by X-
Ligand preparation for docking
ray crystallography to provide a model for structure-basedvirtual screening of commercial chemical compounds in the
The chosen commercial chemical databases are a collection of
ATPase catalytic centre of Pontin. The main criterion for
compounds selected for their high chemical and pharmacological
the database selection was that the molecules were to be available
diversity, as well as their documented bioavailability and safety
for experimental testing, which is why two commercial databases
in humans (according to the manufacturer). The mean Tanimoto
of ligands were used in the virtual screening: the Calbiochem
coefficients are 0.1839 and 0.1265 respectively for Calbiochem
database of Inhibitors® from Merck and the Prestwick Chemical
and Prestwick, which mean a high structural diversity (calculated
Library®. Another advantage was that all of the molecules from
with OpenBabel 2.2.99 and FP2 from Daylight)
these databases display a known activity and are biologically
Compounds from the Calbiochem Database of Kinase
relevant or ‘drug-like', plus their ADME-Tox (absorption,
Inhibitors® were obtained in SD (structure file) format from
distribution, metabolism, excretion and toxicology) data are
the Merck Chemicals website (catalogue numbers are 539743,
documented. The dockings were conducted with Vina then
539744, 539745 and 539746). The three-dimensional structures
the compounds were rescored with DrugScore and Xscore
in MOL2 format were generated automatically using Catalyst in
and the top-scoring ligands were selected via consensus
Discovery Studio via the Prepare Ligands protocol. All
parameters were turned to False, except for Change Ionization,
Finally, priority compounds identified via virtual screening in
the maximum pH was set to 8.5 and the minimum to 6.5 in order
silico were tested in the enzymatic assay in vitro for their activity
to generate different protonation states. The structures obtained
against the ATPase activity of human recombinant Pontin, then in
were then filtered with DBfilter 2.2.8 (a drug-like analyser for
cultured tumour cells for their anti-proliferative effect.
chemical library, distributed by the author S.-H. Wang, 2005).
Molecules with a molecular mass lower than 200 Da or higherthan 800 Da were rejected, as well as molecules with more than ten
rotatable bonds. Only standard atoms such as hydrogen, carbon,nitrogen, oxygen, fluorine, chlorine, bromine, iodine, phosphorus
or sulfur were allowed. All compounds in MOL2 format were
translated into PDBQT files suitable for docking with the script
Selected molecules were from Merck Bioscience and Prestwick.
prepare_ligand4.py from MGLTools 1.5.4
Protease inhibitor tablets were from Roche. The pET21-N-ter-
The same treatment was applied to the compounds from the
His6-TIP49 plasmid was a gift from Dr I.R. Tsaneva (Structural
Prestwick Chemical Library®
and Molecular Biology Group, University College London,
= 26) available on request in SD format. The
London, U.K.). Superdex G 200 HR column was from GE
structures and PDBQT files were automatically generated using
Healthcare. Ni-NTA (Ni2 + -nitrilotriacetate) superflow cartridge
Discovery Studio and the prepare_ligand4.py script.
was from Qiagen. Rottlerin, PTP1B (protein tyrosine phospha-
A total of 29 compounds from the French Chimioth eque Na-
tase 1B) inhibitor {3-(3,5-dibromo-4-hydroxy-benzoyl)-2-ethyl-
tionale were visually selected for experimental evaluation of their
inhibition potential of Pontin ATPase. Because of a non-disclosure
amide} and Akt1/2 inhibitor {1,3-dihydro-1-[1-({4-(6-phenyl-
agreement, we cannot display these structures, but the identific-
ations are available from J.D. upon request. These molecules
yl]-2H-benzimidazol-2-one} were from Calbiochem. Pran-
were manually constructed in Discovery Studio, and Dreiding
Minimize was used to reach a low-energy conformation. As
phenylbutoxy)benzamide} was from Prestwick. Imidazole buffer
described for the compounds from Calbiochem, the PDBQT input
solution and other reagents were from Sigma.
files were automatically prepared with the prepare_ligand4.py
The oligonucleotides used in the present study were (under-
script. These compounds were incorporated into the Calbiochem
lined bases are complementary): single strand (43-mer), 5�-GC-
database and used to evaluate the scoring functions on their ability
to discriminate decoys from potential actives.
� The Authors Journal compilation c
� 2012 Biochemical Society
A combined approach to select Pontin inhibitors
Charges automatically assigned to the ligands during the
Prepare Ligands protocol are not equivalent to the ones assigned
Human recombinant Pontin was purified from 2 litres of
via Dreiding Minimize, but these charges are irrelevant, since Vina
Escherichia coli BL21 culture essentially as described in in
and all the scoring functions used in the present study recalculate
1–2 working days, with a two-step chromatographic procedure.
their own partial charges for the ligands during calculation.
Briefly, following 3 h of induction at 20 ◦C with 1 mM IPTG
(isopropyl β-D-thiogalactopyranoside), cells were lysed in 40 ml
Docking and scoring
of buffer A containing 20 mM Tris/HCl (pH 7.5), 300 mM NaCl,
Autodock Vina 1.0.2 was used for all dockings in the
10 % (v/v) glycerol, 1 mM PMSF, 1 mM 2-mercaptoethanol, pro-
present study. Vina was derived from Autodock, but it achieves
tease inhibitor tablet, 0.5 % Nonidet P40 and 20 mg/ml lysozyme.
improvements in speed and accuracy over the latest release of
Upon incubation for 15 min, the lysate was sonicated extensively
Autodock (Autodock4 Besides, Vina calculations are run
and cleared by centrifugation at 45 000 g for 15 min. The
directly from the command line and automatically take advantage
supernatant was loaded on to a 5 ml Ni-NTA Superflow cartridge
of multiple cores The ligands were docked using Vina, and
column and washed with buffer B containing 20 mM Tris/HCl
then rescored using the stand-alone programs DrugScore
(pH 7.5), 20 mM imidazole, 300 mM NaCl, 10 % (v/v) glycerol
and Xscore. For each ligand in the database, five docking
and 1 mM 2-mercaptoethanol. The eluted fractions collected upon
experiments were conducted, each of them generating up to nine
a 250 mM step with buffer B containing 500 mM imidazole were
poses. The results from the five experiments were averaged, and
then purified on to a Superdex G200 (1 cm×30 cm) in buffer
the compounds were then ranked on the basis of their average
C (buffer B without imidazole). Fractions were analysed by
scores for each individual scoring function or sorted via consensus
SDS/PAGE (12 % gels), pooled and dialysed overnight in buffer
C containing 50 % glycerol, then stored at − 80 ◦C.
Rigid dimer
The ligands were considered flexible while the protein was held
The hydrolysis of ATP into ADP and Pi was measured by the
rigid. The docking grid was designed in order to include the
change in absorbance of the dye Malachite Green in the presence
whole cavity surrounding ADP in the crystallographic structure
of phosphomolybdate complexes using the PiColorLock Gold
plus a margin of at least 3 Å (1 Å = 0.1 nm) in all directions.
assay from Gentaur.
The resulting dimensions of the box were 20 Å×22 Å×20 Å.
The enzymatic reactions were performed in a final volume
A total of 74 amino acids belonging to the first subunit and six
of 100 μl at 37 ◦C. The assay contained a final concentration of
amino acids belonging to the second subunit are fully or partially
20 mM Tris/acetate buffer (pH 7.5), 10 mM magnesium acetate,
located inside the box. The parameters were kept at their default
0.5 mM 2-mercaptoethanol, 100 pmol of DNA, 10 % (v/v)
value. Each docking was conducted five times with five fixed seed
DMSO and catalytic amounts of Pontin. Various serial dilutions
numbers that were used for all ligands. The scores obtained for
of inhibitors dissolved in DMSO were added to a final volume of
each function are the averages of these five experiments.
80 μl. The reactions were started by the addition of 20 μlof the required final concentration of ATP, and its hydrolysis
into ADP and Pi was detected by the colorimetric assay. Thereaction rates were measured by taking 20 μl aliquots at several
The system and parameters were similar to the rigid dimer, except
times and distributed into 96-well plates each containing 20 μl
that 11 torsions from four different residues were considered
of Picolor Gold and 60 μl of water for at least 10 min at
rotatable during docking.
room temperature (22 ◦C). The stain was then stabilized with
10 μl of ‘Stabilizer' for 30 min at room temperature. The A620
was measured using a spectrophotometer (Labsystems MultiskanBichromatic) and correlated against a standard curve of P
The blind docking was conducted on a monomer of Pontin. The
slopes were determined, thus providing the reaction rates. All
parameters were kept at their default values once again, but the
experiments were carried out in triplicate.
search space was extended considerably to encompass the whole
The reproducibility of the assay was tested by measuring the Z'
protein. The box dimensions were 58 Å×76 Å×70 Å.
value. This was done by measuring the reaction rates over 30 min
The docking output PDBQT-formatted files were translated into
with 10 μM Pontin and 1 mM ATP. The Z' value was deduced
PDB with in-house scripts and non-polar hydrogens were added
from 48 reaction rates and found to be equal to 0.7. This value
in MOL2 format with OpenBabel 2.2.3 All poses generated
indicates the good reproducibility of the assay since accepted
by Vina were rescored using scoring functions from DrugScore
values of Z' lie between 0.5 and 1
and Xscore, resulting in ten different scores for each pose.
The Consensus scoring protocol from Discovery Studio was
used to create a list of priority compounds.
Molecules were dissolved in DMSO. The final amount of DMSO
was 10 % in the assays and we checked that it did not interfere
Manual checking of the three-dimensional structures of the
with the reaction rates. We also checked that at the highest
ligands was performed on the top-scoring molecules only, after
tested concentration of 50 μM, none of the molecules interfered
calculations. The automatic generation of the structures had
with the assay in the presence of Pi (results not shown). For
eventually produced some errors, mostly on heterocyclic aromatic
each concentration of inhibitor, the reaction rates were measured
features. Therefore all molecules included in the best consensus
upon addition of 50 μM ATP. The IC50 (half maximal inhibitory
for both databases were checked visually for errors, corrected
concentration) was determined from two enzyme purifications.
and redocked if needed before the identification of compounds
The reported IC50 values are the means for three independent
selected for experimental screening.
� The Authors Journal compilation c
� 2012 Biochemical Society
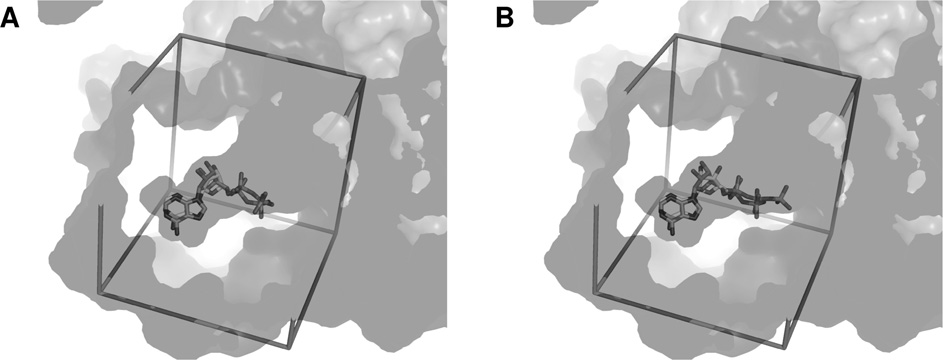
J. Elkaim and others
Docking of ADP and ATP
(A) Superimposition of crystallized ADP from PDB code 2C9O and docked ADP into the dimer of Pontin. The active site is represented as transparency, the search space box is represented as sticks.
(B) Similar representation with docked ATP.
Cell proliferation experiments
phosphate chain of ADP was stabilized by a network of eighthydrogen bonds coming from six different residues, whereas that
The human hepatic tumour cell lines HuH7 and Hep3B were used.
of ATP could create up to 13 hydrogen bonds with eight residues,
Cells were seeded at an initial density of 2500 cells/well in 96-
including five out of the six found in the case of ADP.
well plates in DMEM (Dulbecco's modified Eagle's medium) with10 % (v/v) fetal bovine serum. On the following day, they weretreated with dilutions of test molecules in DMSO. The DMSO
Constitution of priority lists of compounds by consensus scoring
concentration was adjusted to 1 % in every well. After 4 days, cell
Considering the very few experimental results, the approach based
numbers were estimated colorimetrically at 492 nm with the Cell-
on individual scoring function was very likely to produce a large
Titer 96 Aqueous One Solution Cell Proliferation Assay {MTS [3-
number of misranks. In this context, a consensus scoring strategy
seemed a reasonable choice that could result in a reduction in
sulfophenyl)-2H-tetrazolium] assay} from Promega. The growth
the number of false negatives when selecting a limited number of
index was calculated using eqn (1):
molecules for experimental testing
DrugScoreCSD completely failed to eliminate the decoys from the
top scorers, whereas Vina, DrugScorePDB and Xscore provided farbetter results. In conjunction with this, a very good consensus
where D4T refers to the D492 with the test molecule at day 4, D0
was obtained with HPscore from Xscore and SURF from
is the D at day 0, and D4DMSO is the D at day 4 with DMSO alone.
DrugScore, which provided a large number of common top-scoring compounds including only one decoy, and adding Vinainto the consensus allowed us to eliminate the last decoy in the
RESULTS AND DISCUSSION
top list. This improvement was followed by a significant decreasein the number of compounds in the intersection, but considering
that the dockings were made with Vina and that its scoring
As mentioned above, no Pontin inhibitor had ever been reported
function was used to generate the poses of the ligands, we
in the literature, but according to the enzymatic assay described
decided that the Vina scoring function should be included in the
below, an initial test on analogues of ATP had allowed us to
consensus (see the Supplementary Online Data at
identify 29 inactive compounds (results not shown). The ability
for a detailed descrip-
to discriminate these decoys presented by our scoring functions
tion of the virtual screening results).
either independently or in conjunction was used as a tool in order
The consensuses carried out with these three independent
to evaluate the correctness of our results. At first, 900 compounds
scoring functions on the Calbiochem database led to the identi-
from the Calbiochem database plus the 29 decoys from the French
fication of 22 priority compounds ranking in the top 15 %
Chimioth eque Nationale were used as a training set. Then, 1299
for the three functions. Ten of them, i.e. approximately 1 % of
compounds from Prestwick were screened with regard to the
the complete database, were randomly selected for experimental
results obtained with the Calbiochem training set.
screening. The same procedure was applied to the 1299
Before the docking of the databases, ATP and ADP were docked
compounds from the Prestwick Chemical Library®, leading to
into the active site to evaluate the quality of the model. The
the prioritization of 23 new molecules and approximately the
resulting poses were very similar to crystallized ADP
same percentage, namely 15 compounds, were chosen for testing.
In particular, the adenine groups and the ribose of both moleculeswere superimposed on that of crystallized ADP, whereas only the
Design of a versatile ATPase assay to measure Pontin activity
highly flexible phosphate chain of ATP was notably displacedin some of the poses. At least 15 residues were involved in
Human recombinant Pontin was purified via chromatography and
hydrophobic contacts with both ADP and ATP. In addition, the
the fractions eluted yielded various amounts of dodecameric,
� The Authors Journal compilation c
� 2012 Biochemical Society

A combined approach to select Pontin inhibitors
stimulation of the reaction rate was observed as shown in(P < 0.01). The effects of a single-stranded DNAof 43-mer, a hairpin of 44-mer and a perfect duplex DNA of64-mer were studied. No significant differences were foundwhichever polynucleotide was used (results not shown). TheATP-hydrolysis reaction rate stimulation in the presence of DNAcorrelates with the presence of a polynucleotide-binding sitedetected by gel-shift assays with Pontin This activation byDNA is controversial in the literature, since it has been reportedby many groups but could not be detected by othersHowever, the presence of DNA was essential in our assayto detect a sufficient activity in order to measure the IC50 of theligands.
In order to test for potential inhibitors, the ATP concentration
corresponding to the initial velocity was determined. Catalysisfollowed an asymptotic curve, allowing the determination of theKm (Michaelis constant) with the Michaelis–Menten equation(eqn 2):
V = Vmax[S]
Using the double-reciprocal plot described by Lineweaver–
Burk, the Km was found to be equal to 50 μM ( +
− 15 μM) as shown
in Thus the inhibition curves of the compounds wereobtained using an ATP concentration of 50 μM, and the IC50 aswell as the resulting inhibition constant Ki could be calculatedthanks to the Cheng–Prusoff relationship (eqn 3):
with [ATP]/K =
Enzymatic assays of Pontin ATPase activity
(A) SDS/PAGE of 5 μg of the monomeric Pontin fraction purified by chromatography on
Superdex G200, and stained with Coomassie Brilliant Blue. Molecular masses are indicated
in kDa. (B) Relationship between Pi concentration and A 620 (‘OD 620 nm'). Increasing
All compounds were initially tested at a 50 μM concentration.
concentrations of Pi were used with the Malachite Green assay, as described in the Experimental
When no significant variation of the reaction rates were measured
section. (C) Reaction rates according to the concentration of Pontin. The reported rates were
determined for each concentration of Pontin during a 40 min incubation at 37 ◦C. Aliquots of
(i.e. approximately 30 %), we considered the compounds as
20 μl were taken every 10 min and the release of P
i was measured as in (B). (D) Effect
of 10 μM DNA (43-mer) on the reaction rates in the presence of 2.5 μM Pontin and 2 mM ATP.
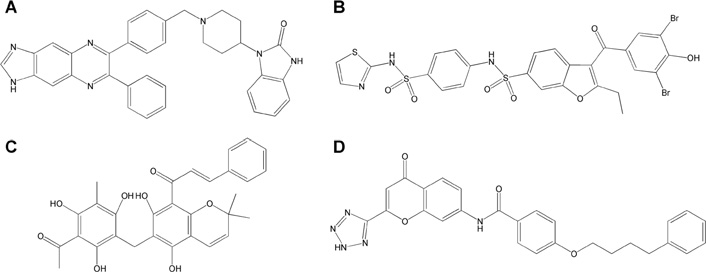
Akt1/2 inhibitor inhibited Pontin ATPase activity with an IC50
The presence of DNA significantly increased the reaction rate (ANOVA, P < 0.001). Similar
of 24 μM This compound was reported to inhibit
results were found with the other oligonucleotides described in the Experimental section. (E)
Akt1 preferentially (IC
Lineweaver–Burk representation of the reaction rates according to the ATP concentration in the
50 of 58 nM) with also a good potency
assays. The concentration of Pontin was 2 μM. Results in (B)–(D) are means +
− S.D. for three
50 of 210 nM) and less against Akt3 (IC50 of
2.1 μM).
A non-competitive specific inhibitor of PTP1B presented an
IC50 of 15 μM compared with 4 μM for its primary
hexameric and monomeric forms of Pontin as shown by
elution volume and SDS/PAGE. When tested by the ATPase
Rottlerin inhibited Pontin activity with an IC50 of 10 μM
assay described below, only the purified monomeric fraction
This molecule was also reported to be a reversible
proved to be active, therefore all enzymatic
inhibitor of the PKC family, although this has been questioned and
experiments were conducted on the monomeric fraction.
it may also inhibit several MAPK family members Using
A Malachite Green-derived assay that quantifies the green com-
the Cheng–Prusoff equation (eqn 3 the Ki values for these
plex formed between Malachite Green, molybdate and free Pi was
three molecules were determined and found to be 12.1, 7.5 and
used to measure the enzyme activity in 96-well plates. As shown
5.1 μM respectively.
on A620 was proportional to the Pi concentration. The
From the 15 compounds prioritized in the Prestwick Chemical
reaction rates were proportional to the enzyme concentration, and
Library®, only Pranlukast, a potent and specific competitive
did not display apparent co-operativity
antagonist of the cysteinyl leukotriene 1 receptor, inhibited the
Almost no variation in the reaction rate of the ATPase
ATPase reaction with an IC50 of 13 μM thus with a
catalysis could be detected in assays performed in the absence
Ki of 6.5 μM.
of oligonucleotides. In contrast, upon addition of an excess of
The chemical structures of these compounds are shown in
single- or double-stranded DNA ([DNA]/[E] = 1–10), a 3-fold
� The Authors Journal compilation c
� 2012 Biochemical Society

J. Elkaim and others
In all cases, an aromatic cycle lay in a position close to that
of the adenine. Furthermore, Pranlukast and the PTP1B inhibitorpresented a hydrogen-bond acceptor group superimposed on the
γ -phosphate of ATP, and created hydrogen-bonding with Gly73similar to both ATP and ADP. In contrast, the pose of the Akt1/2inhibitor prevented all hydrogen-bonding with this side of thecavity. Rottlerin was the only ligand to form a hydrogen bondwith Arg404 from sensor 2, as in the case of ATP.
As opposed to ADP or ATP, all compounds created interactions
with the second area of the cavity. This included hydrophobiccontacts with the second subunit, through Asp353 for Rottlerin,as well as Asp356 for all ligands. In addition, Pranlukast and thePTP1B inhibitor made electrostatic interactions with the arginine‘finger', i.e. Arg357 from the second subunit. The latter evenaccepted two hydrogen bonds between its sulfonamide and theguanidine group from the arginine finger.
A flexible docking was also conducted on the dimer, meaning
that the rotations on the side chains of the residues located in asphere of 5 Å around the centre of mass of ADP were authorizedduring docking. This method aims to render the rearrangementsinduced by the binding of a ligand into the structure of the active
Identification of four inhibitors
site (induced fit), but the major drawback is the calculation time,which increases dramatically with the number of torsions allowed.
Determination of the IC50 of the Akt1/2 inhibitor (A), the PTP1B inhibitor (B), Rottlerin (C)
and Pranlukast (D) with 50 μM ATP and 4 μM Pontin. Results are means +
In the model, the active site was completely buried and highly
− S.D. for four
experiments. Some error bars cannot be seen because they were too small.
constrained. The authorized movements on the side chains werethus very limited. As expected, we have observed no noticeablemodifications in the results obtained or in the poses compared
Binding pose analysis
with those obtained with the non-flexible dimer.
The observation of the active-site topology led to the distinctionof two spaces. The flattest part of the cavity was occupied by ADPand ATP, whereas the other side, which includes all residues from
Type of enzyme inhibition
the second subunit, was left empty. The best docking poses for allactive compounds shared common features with ADP and ATP
The kind of inhibition presented by these compounds was
determined by measuring the rates of ATP hydrolysis with
Out of the 17 residues involved in the binding mode of ADP, ten
the Malachite Green assay, using five concentrations of ATP,
were also implicated in the binding of all inhibitors. The number of
ranging from the Km (50 μM) to 1 mM, together with increasing
residues common to the binding mode of ADP and to the ligands
concentrations of inhibitors.
respectively ranged from 11 for Rottlerin, to 13 for Pranlukast and
The double-reciprocal plots showed that Rottlerin
14 for Akt and PTP1B inhibitors. Furthermore, various residues
was the only ligand that inhibited the ATPase activity
from the Walker A domain (residues 70–79) were involved in the
competitively. In the experiments using Pranlukast and the PTP1B
binding mode of all ligands as well as ADP and ATP, with at least
inhibitor, both the Vmax and the Km for ATP hydrolysis varied upon
four different residues out of ten in close proximity to the ligands.
addition of the inhibitors, revealing a mixed or uncompetitive
As for the Walker B domain (residues 302–305), all inhibitors
inhibition. The analysis of the results obtained with the Akt1/2
and ATP interacted with Asp302 only, whereas ADP did not make
inhibitor revealed a more surprising inhibition profile since the
contact with it at all. This residue is crucial for the hydrolysis
double-reciprocal plots indicated that the − 1/Km value was
of ATP, as shown by the lack of ATPase activity for the D302N
identical with the one measured for ATP, proving that the inhibitor
did not compete with the ATP-binding site.
Chemical structures of the four active compounds
(A) Akt1/2 inhibitor. (B) PTP1B inhibitor. (C) Rottlerin. (D) Pranlukast.
� The Authors Journal compilation c
� 2012 Biochemical Society

A combined approach to select Pontin inhibitors
Docking poses in the dimer of Pontin
(A) The best docking pose for the Akt1/2 inhibitor is shown as sticks. The active site is represented as transparency with surfaces of both subunits of Pontin shown in different shades of grey.
Important domains from the ATP-binding site are shown as lines with essential residues as sticks. From the first subunit, the Walker A domain is in front of the ligands, the Walker B domain is at the
right side of the image, with Asp302 highlighted, and the sensor 2 is at the back of the cavity, with Arg404 highlighted. From the second subunit, the arginine finger (Arg357) is shown as sticks. The
search space box is represented in sticks. (B–D) Similar representation of the PTP1B inhibitor (B), Rottlerin (C) and Pranlukast (D).
The compounds tested experimentally had been selected via
on the monomer, the ligands had the freedom not only to explore
structure-based virtual screening. According to the literature,
the surface surrounding the cavity that was covered up by the
the docking experiments were conducted by targeting the ATP-
second subunit in the dimer, but also to bind everywhere on
binding site and we therefore expected the inhibitors
the surface of whole protein.
that were identified to bind competitively to the ATP-binding site
In the case of the Akt1/2 inhibitor, similar results were obtained
of Pontin. Yet, among four inhibitors, only Rottlerin appeared to
either using Pontin alone or in complex with ADP and ATP.
be competitive. Pranlukast and the PTP1B inhibitor presented a
Except for a few poses halfway inside the active site when Pontin
mixed or uncompetitive profile and, in the case of the Akt inhibitor,
was used alone, or situated at the entrance of the active site when
the double-reciprocal plot indicated a non-competitive inhibitor.
ATP was present, almost all of the poses observed were located
In order to explore other potential binding modes, additional
in a groove at the junction between domain I and domain II of the
docking experiments were conducted on the monomer of Pontin,
protein These poses were too far from the active site
in the presence or in the absence of ATP and ADP, considering
to interfere directly with the binding of ATP, but they could be
a search space that encompassed the whole protein (blind
interacting with the nucleotide-binding site. Actually, domain II
docking). In the case of a tandem dimer docking, the search
is the seat of the DNA-binding site, and since we have shown that
box contained only the binding site of crystallized ADP, which
the presence of DNA considerably influenced the reaction rate,
was completely buried since the second monomer was closing
it is possible that the Akt1/2 inhibitor interferes with this feature.
the cavity, contributing to the shape of the active site. This
As for Pranlukast, the blind docking has shown that when
subunit, even if it did not create interactions with the compounds,
ATP was complexed with Pontin, a binding mode located at the
constrained them into a closed area, playing a key role in the
entrance of the cavity was clearly favoured This
ligand-binding modes. In contrast, in the case of the blind docking
pose could prevent ATP from being hydrolysed and does not
� The Authors Journal compilation c
� 2012 Biochemical Society
J. Elkaim and others
Types of enzyme inhibition
(A) Double-reciprocal plots (Lineweaver–Burk plots) of the reaction rates with ATP alone, and increasing concentrations of Rottlerin. When no variation of 1/[ATP] is observed, the inhibitor is
non-competitive and does not modify the binding of ATP. In contrast, when no variation of 1/V max is observed, the ligand is competitive and disrupts ATP binding to the protein. (B–D) Similar plots
for Pranlukast (B), PTP1B inhibitor (C) and Akt1/2 inhibitor (D). Results are means for two independent experiments performed four times.
Docking poses obtained via blind docking
(A) The best docking pose for the Akt1/2 inhibitor is shown as sticks. The monomer of Pontin is represented as transparency and cartoons, with domains I, II and III coloured different shades of
grey. The inhibitor lies in a groove between domain I and domain II, in close proximity to the DNA-binding site. (B) Similar representation for Pranlukast. The inhibitor is located at the entrance
of the catalytic site.
leave sufficient space for ADP to get out. It is also incompatible
halfway inside the active site, which was contradictory to the
with the existence of a tandem dimer similar to that observed
experimental data. These results are consistent with the hypothesis
in crystallography as Pranlukast would create clashes with
of an uncompetitive inhibition, which implies that the enzyme is
several residues from the second subunit. In contrast, no clear
complexed by its substrate before the binding of the inhibitor.
pose preference was observed when Pontin was complexed with
The binding of ATP would thus enable Pranlukast to bind to the
ADP, and the best poses obtained with Pontin alone were located
protein and to inhibit its enzymatic activity.
� The Authors Journal compilation c
� 2012 Biochemical Society
A combined approach to select Pontin inhibitors
The blind docking of the PTP1B inhibitor did not reveal
such a clear-cut profile. The complexation of ATP did slightlyfavour the pose at the entrance of the cavity as in the case ofPranlukast, but numerous other poses were observed as well,and it was hard to define which pose was the most likely to beobserved.
Taken together, these experiments would warrant further
structural and biochemical studies due to the complexity of Pontin.
The competitive inhibition observed with Rottlerin indicated apure competition with the ATP-binding site which confirmedthe in silico antagonists selection. Nonetheless, the mixed oruncompetitive inhibition measured with the PTP1B inhibitor andPranlukast, and the non-competitive nature of the Akt inhibitor,suggest indirect interferences with the ATPase activity of Pontin,either by blocking the hydrolysis of ATP or the exit of ADP,or through interactions with the DNA-binding site. Thereforethe modulation of the hydrolysis shown upon addition of DNAmay be an important parameter in these processes. Actually, asshown by Mezard et al. and Rottbauer et al. DNA-binding and ATPase activities are tightly coupled. Moreover, thecatalytic site is formed by two subunits and we can assume thatthe arginine finger may indirectly modulate the catalytic activityupon inhibitor binding A similar inference may be suggestedby the observations of Zhang and Wigley and Moffitt et al.
pointing to intersubunit interactions.
Effects of the inhibitors on proliferation of cultured cell lines
The hepatic tumour cell lines HuH7 and Hep3B were grown
Cell proliferation assays
in the presence of these compounds for 4 days. As can
Effects of Rottlerin (A and B), Akt inhibitor (C and D) and Pranlukast (E and F) on the growth
be seen in Rottlerin had a strong anti-proliferative
of HuH7 (A, C and E) and Hep3B (B, D and F) hepatocarcinoma cell lines. Cells were grown in
effect on both cell lines. It was toxic at concentrations higher
the presence of various concentrations of the inhibitors (indicated in μM). Cell numbers were
than 5 μM for HuH7 and 1 μM for Hep3B, as was evident
estimated 4 days later using the MTS assay, and the growth index was calculated as described
from cell numbers falling below the day 0 values. The Akt
in the Experimental section. Results are means for two independent experiments performed withfive replicates each.
inhibitor behaved similarly, although it was toxic only at higherconcentrations. Pranlukast was not toxic in the concentrationrange tested, and decreased cell proliferation of both cell
lines dose-dependently. Calculated IC50 values in HuH7 andHep3B cells were respectively 0.57 and 0.25 μM for Rottlerin,
Using in silico and in vitro complementary approaches, the present
4.0 and 3.5 μM for Akt1/2 inhibitor, and 63.7 and 34.3 μM
paper discloses the first identification of inhibitors of the ATPase
for Pranlukast. Finally, the PTP1B inhibitor did not show
activity of Pontin, an activity required for several biological
any growth inhibition or cytotoxicity at concentrations up
to 100 μM.
The first step involved structural modelling of the ATP-binding
Altogether, three out of four molecules reduced cell numbers as
centre, and virtual screening of approximately 2200 commercial
seen with Pontin silencing suggesting that they may indeed
compounds using Vina. Thorough rescoring of the poses with
target Pontin within cells. Because of their cognate targets, it
Xscore and DrugScore, using 29 decoys as a negative calibration
was expected that Rottlerin and the Akt inhibitor would have
set, allowed the selection of efficient scoring functions for
such an effect. Since these molecules are likely to act at the
this system. Various consensuses using these functions were
same time on those targets and on Pontin, it is difficult to strictly
compared, and the intersection of Vina, SURF score from
compare their in vitro and in vivo IC50 values and we can thus
DrugScore and HPscore from Xscore scores appeared to provide
only conclude that their effects are compatible with an effect
the best results. With regard to these results, this consensus
on Pontin. PTP1B inhibition can lead either to increased or
was used to prioritize 25 compounds for experimental testing.
reduced cell proliferation, depending on the context In our
This virtual screening strategy proved efficient, since post-
hands, the PTP1B antagonist had no effect on the growth of
experimental analysis of the results indicated that the consensus
the hepatocellular carcinoma cell lines tested. It may be that
scoring used to discriminate the compounds was the only one
access of the inhibitor to Pontin is hampered in vivo, either
that allowed the selection of the four active molecules. Similarly,
because of internalization or solubility issues, or because the
none of the single functions had detected all four ligands in their
in vivo conformation of the target prevents the inhibitor from
top-scoring lists.
accessing the catalytic centre. Finally, Pranlukast, an antagonist of
The enzymatic testing of chemicals selected by docking
the leukotriene receptor, reduced cell proliferation with a similar
with a Malachite Green assay was made possible because of
potency as for Pontin ATPase activity inhibition. This is of special
improvements in the procedure, and in particular the addition of
interest since, in contrast with the other three molecules for which
polynucleotides that increased the reaction rates severalfold. Four
primary targets are nucleotide-using enzymes, Pranlukast is a
ligands displayed an inhibition constant in the micromolar range.
competitive antagonist of a non-nucleotide molecule believed to
The colorimetric assay described in the present paper could also
act outside the plasma membrane.
be used for high-throughput screenings, because of its sensitivity
� The Authors Journal compilation c
� 2012 Biochemical Society
J. Elkaim and others
and rapidity compared with the current time-consuming use of
11 Jin, J., Cai, Y., Yao, T., Gottschalk, A. J., Florens, L., Swanson, S. K., Gutierrez, J. L.,
radiolabelled ATP.
Coleman, M. K., Workman, J. L., Mushegian, A. et al. (2005) A mammalian chromatin
Finally, three of the four compounds tested reduced cell growth
remodeling complex with similarities to the yeast INO80 complex. J. Biol. Chem. 280,
at concentrations close to those inhibiting the ATPase activity of
12 Shen, X., Mizuguchi, G., Hamiche, A. and Wu, C. (2000) A chromatin remodelling
Pontin in vitro. Whether their anti-proliferative action is due to
complex involved in transcription and DNA processing. Nature 406, 541–544
Pontin antagonism will require further study.
13 Jha, S., Shibata, E. and Dutta, A. (2008) Human Rvb1/Tip49 is required for the histone
acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on
H2AX after DNA damage. Mol. Cell. Biol. 28, 2690–2700
14 Gallant, P. (2007) Control of transcription by Pontin and Reptin. Trends Cell. Biol. 17,
Judith Elkaim designed, performed and analysed the virtual screening experiments. Michel
15 Bauer, A., Chauvet, S., Huber, O., Usseglio, F., Rothbacher, U., Aragnol, D., Kemler, R. and
Castroviejo provided invaluable help in protein purification. Driss Bennani designed and
Pradel, J. (2000) Pontin52 and reptin52 function as antagonistic regulators of β-catenin
wrote specific scripts for the virtual screening analysis. Said Taouji adapted and performed
signalling activity. EMBO J. 19, 6121–6130
the robotized biochemical screening, analysed the results and prepared the corresponding
16 Wood, M. A., McMahon, S. B. and Cole, M. D. (2000) An ATPase/helicase complex is an
Figures. Nathalie Allain and Patrick Lestienne performed the protein purification, and
essential cofactor for oncogenic transformation by c-Myc. Mol. Cell 5, 321–330
participated in the biochemical testing, analysis and Figure preparation. Michel Laguerre
17 Watkins, N. J., Dickmanns, A. and Luhrmann, R. (2002) Conserved stem II of the box C/D
prepared the three-dimensional structure of Pontin and designed the virtual screening
motif is essential for nucleolar localization and is required, along with the 15.5K protein,
experiments. Patrick Lestienne designed the overall biochemical experiment and realized
for the hierarchical assembly of the box C/D snoRNP. Mol. Cell. Biol. 22, 8342–8352
the enzymatic assay. Jean Dessolin designed the virtual screening experiments and
18 Boulon, S., Marmier-Gourrier, N., Pradet-Balade, B., Wurth, L., Verheggen, C., Jady, B. E.,
analysed the results. Judith Elkaim, Said Taouji, Michel Laguerre, Jean Rosenbaum,
Rothe, B., Pescia, C., Robert, M. C., Kiss, T. et al. (2008) The Hsp90 chaperone controls
Patrick Lestienne and Jean Dessolin discussed the data. Judith Elkaim, Jean Rosenbaum,
the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 180, 579–595
Patrick Lestienne and Jean Dessolin wrote the paper.
19 Venteicher, A. S., Meng, Z., Mason, P. J., Veenstra, T. D. and Artandi, S. E. (2008)
Identification of ATPases pontin and reptin as telomerase components essential for
holoenzyme assembly. Cell 132, 945–957
20 Haurie, V., Menard, L., Nicou, A., Touriol, C., Metzler, P., Fernandez, J., Taras, D.,
Lestienne, P., Balabaud, C., Bioulac-Sage, P. et al. (2009) Adenosine triphosphatase
We thank the French Chimioth eque Nationale for initial studies. We gratefully acknowledge
pontin is overexpressed in hepatocellular carcinoma and coregulated with reptin through
Dr I.R. Tsaneva for the gift of the plasmid and for helpful and friendly discussion. Weexpress our warmest thanks to Dr S.-H. Wang (Graduate Institute of Pharmaceutical
a new posttranslational mechanism. Hepatology 50, 1871–1883
Chemistry, China Medical University, Taiwan) for providing DBfilter and Dr M.L. Jung
21 Rousseau, B., Menard, L., Haurie, V., Taras, D., Blanc, J., Moreau-Gaudry, F., Metzler, P.,
from Prestwick.
Hugues, M., Boyault, S., Lemiere, S. et al. (2007) Overexpression and role of the ATPase
and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology
46, 1108–1118
22 Menard, L., Taras, D., Grigoletto, A., Haurie, V., Nicou, A., Dugot-Senant, N., Costet, P.,
Rousseau, B. and Rosenbaum, J. (2010) In vivo silencing of Reptin blocks the
This project was supported by grants from Ligue Nationale Contre le Cancer, Association
progression of human hepatocellular carcinoma in xenografts and is associated with
pour la Recherche sur le Cancer and Institut National du Cancer [grant number PLBIO10-
replicative senescence. J. Hepatol. 52, 681–689
155]. We thank the MENRT (Ministere de l'Education Nationale de la Recherche et de
23 King, T. H., Decatur, W. A., Bertrand, E., Maxwell, E. S. and Fournier, M. J. (2001) A
Technologie), CNRS (Centre National de la Recherche Scientifique) and INSERM (Institut
well-connected and conserved nucleoplasmic helicase is required for production of box
National de la Sant´e et de la Recherche M´edicale) for their support.
C/D and H/ACA snoRNAs and localization of snoRNP proteins. Mol. Cell. Biol. 21,
7731–7746
24 Lim, C. R., Kimata, Y., Ohdate, H., Kokubo, T., Kikuchi, N., Horigome, T. and Kohno, K.
(2000) The Saccharomyces cerevisiae RuvB-like protein, Tih2p, is required for cell cycle
progression and RNA polymerase II-directed transcription. J. Biol. Chem. 275,
1 Jonsson, Z. O., Dhar, S. K., Narlikar, G. J., Auty, R., Wagle, N., Pellman, D., Pratt, R. E.,
Kingston, R. and Dutta, A. (2001) Rvb1p and Rvb2p are essential components of a
25 Feng, Y., Lee, N. and Fearon, E. R. (2003) TIP49 regulates β-catenin-mediated neoplastic
chromatin remodeling complex that regulates transcription of over 5 % of yeast genes. J.
transformation and T-cell factor target gene induction via effects on chromatin
Biol. Chem. 276, 16279–16288
remodeling. Cancer Res. 63, 8726–8734
2 Mezard, C., Davies, A. A., Stasiak, A. and West, S. C. (1997) Biochemical properties of
26 Dugan, K. A., Wood, M. A. and Cole, M. D. (2002) TIP49, but not TRRAP, modulates
the RuvBD113N: a mutation in helicase motif II of the RuvB hexamer affects DNA binding
c-Myc and E2F1 dependent apoptosis. Oncogene 21, 5835–5843
and ATPase activities. J. Mol. Biol. 271, 704–717
27 Qiu, X. B., Lin, Y. L., Thome, K. C., Pian, P., Schlegel, B. P., Weremowicz, S., Parvin, J. D.
and Dutta, A. (1998) An eukaryotic RuvB-like protein (RUVBL1) essential for growth. J.
3 Matias, P. M., Gorynia, S., Donner, P. and Carrondo, M. A. (2006) Crystal structure of the
Biol. Chem. 273, 27786–27793
human AAA + protein RuvBL1. J. Biol. Chem. 281, 38918–38929
28 Ikura, T., Ogryzko, V. V., Grigoriev, M., Groisman, R., Wang, J., Horikoshi, M., Scully, R.,
4 Ammelburg, M., Frickey, T. and Lupas, A. N. (2006) Classification of AAA + proteins. J.
Qin, J. and Nakatani, Y. (2000) Involvement of the TIP60 histone acetylase complex in
Struct. Biol. 156, 2–11
DNA repair and apoptosis. Cell 102, 463–473
5 Gribun, A., Cheung, K. L., Huen, J., Ortega, J. and Houry, W. A. (2008) Yeast Rvb1 and
29 Choi, J., Heo, K. and An, W. (2009) Cooperative action of TIP48 and TIP49 in H2A.Z
Rvb2 are ATP-dependent DNA helicases that form a heterohexameric complex. J. Mol.
exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 37,
Biol. 376, 1320–1333
6 Puri, T., Wendler, P., Sigala, B., Saibil, H. and Tsaneva, I. R. (2007) Dodecameric structure
30 Trott, O. and Olson, A. J. (2010) AutoDock Vina: improving the speed and accuracy of
and ATPase activity of the human TIP48/TIP49 complex. J. Mol. Biol. 366, 179–192
docking with a new scoring function, efficient optimization, and multithreading. J.
7 Torreira, E., Jha, S., Lopez-Blanco, J. R., Arias-Palomo, E., Chacon, P., Canas, C., Ayora,
Comput. Chem. 31, 455–461
S., Dutta, A. and Llorca, O. (2008) Architecture of the pontin/reptin complex, essential in
31 Velec, H. F. G., Gohlke, H. and Klebe, G. (2005) DrugScoreCSD: knowledge-based scoring
the assembly of several macromolecular complexes. Structure 16, 1511–1520
function derived from small molecule crystal data with superior recognition rate of near
8 Gorynia, S., Bandeiras, T. M., Pinho, F. G., McVey, C. E., Vonrhein, C., Round, A.,
native ligand poses and better affinity prediction. J. Med. Chem. 48, 6296–6303
Svergun, D. I., Donner, P., Matias, P. M. and Carrondo, M. A. (2011) Structural and
32 Gohlke, H., Hendlich, M. and Klebe, G. (2000) Knowledge-based scoring function to
functional insights into a dodecameric molecular machine: the RuvBL1/RuvBL2 complex.
predict protein–ligand interactions. J. Mol. Biol. 295, 337–356
J. Struct. Biol. 176, 279–291
33 Wang, R., Laib, L. and Wang, S. (2002) Further development and validation of empirical
9 Grigoletto, A., Lestienne, P. and Rosenbaum, J. (2011) The multifaceted proteins Reptin
scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol.
and Pontin as major players in cancer. Biochim. Biophys. Acta 1815, 147–157
Des. 16, 11–26
10 Jha, S. and Dutta, A. (2009) RVB1/RVB2: running rings around molecular biology. Mol.
34 O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T. and Hutchison, G.
Cell 34, 521–533
R. (2011) Open Babel: an open chemical toolbox. J. Cheminform. 3, 33
� The Authors Journal compilation c
� 2012 Biochemical Society
A combined approach to select Pontin inhibitors
35 Sanner, M. F. (1999) Python: a programming language for software integration and
43 Makino, Y., Kanemaki, M., Kurokawa, Y., Koji, T. and Tamura, T.-A. (1999) A rat Ruv-B like
development. J. Mol. Graphics Modell. 17, 57–61
Protein, TIP49a, is a germ cell-enriched novel DNA helicase. J. Biol. Chem. 274,
36 Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K. and
Olson, A. J. (1998) Automated docking using a Lamarckian genetic algorithm and an
44 Rottbauer, W., Saurin, A. J., Lickert, H., Shen, X., Burns, C. G., Wo, Z. G., Kemler, R.,
empirical binding free energy function. J. Comput. Chem. 19, 1639–1662
Kingston, R., Wu, C. and Fishman, M. (2002) Reptin and pontin antagonistically regulate
37 Chang, M. W., Ayeni, C., Breuer, S. and Torbett, B. E. (2010) Virtual screening for HIV
heart growth in zebrafish embryos. Cell 111, 661–672
protease inhibitors: a comparison of AutoDock 4 and Vina. PLoS ONE 5, e11955
45 Cheng, Y. and Prusoff, W. H. (1973) Relationship between inhibition constant (K i) and the
38 Guha, R., Howard, M. T., Hutchison, G. R., Murray-Rust, P., Rzepa, H., Steinbeck, C.,
concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic
Wegner, J. K. and Willighagen, E. (2006) The Blue Obelisk: interoperability in chemical
reaction. Biochem. Pharmacol. 22, 3099–3108
informatics. J. Chem. Inf. Model. 46, 991–998
46 Soltoff, S. P. (2007) Rottlerin: an inappropriate and ineffective inhibitor of PKCδ. Trends
39 Zhang, J. H., Chung, T. D. and Oldenburg, K. R. (1999) A simple statistical parameter for
Pharmacol. Sci. 28, 453–458
use in evaluation and validation of high throughput screening assays. J. Biomol. Screen.
47 Hishida, T., Han, Y. W., Fujimoto, S., Iwasaki, H. and Shinagawa, H. (2004) Direct
4, 67–73
evidence that a conserved arginine in RuvB AAA + ATPase acts as an allosteric effector
40 Charifson, P. S., Corkery, J. J., Murcko, M. A. and Walters, W. P. (1999) Consensus
for the ATPase activity of the adjacent subunit in hexamer. Proc. Natl. Acad. Sci. U.S.A.
scoring: a method for obtaining improved hit rates from docking databases of
three-dimensional structures into proteins. J. Med. Chem. 42, 5100–5109
48 Zhang, X. and Wigley, D. B. (2008) The "glutamate switch" provides a link, between
41 Wang, R. and Wang, S. (2001) How does consensus scoring work for virtual library
ATPase activity and ligand binding in AAA + proteins. Nat. Struct. Mol. Biol. 11,
screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 41,
49 Moffitt, J. R., Chemila, Y. R., Aathavan, K., Grimes, S., Jardine, P. J., Anderson, D. L. and
42 Kanemaki, M., Kurokawa, Y., Matsu-ura, T., Makino, Y., Masani, A., Okazaki, K.,
Bustamante, C. (2009) Intersubunit coordination in a homomeric ring ATPase. Nature
Morishita, T. and Tamura, T. A. (1999) TIP49b, a new RuvB-like DNA helicase, is included
in a complex together with another RuvB-like DNA helicase, TIP49a. J. Biol. Chem. 274,
50 Lessard, L., Stuible, M. and Tremblay, M. (2010) The two faces of PTP1B in cancer.
Biochim. Biophys. Acta 1804, 613–619
Received 5 October 2011/23 January 2012; accepted 24 January 2012Published as BJ Immediate Publication 24 January 2012, doi:10.1042/BJ20111779
� The Authors Journal compilation c
� 2012 Biochemical Society
Biochem. J. (2012) 443, 549–559 (Printed in Great Britain)
SUPPLEMENTARY ONLINE DATA
First identification of small-molecule inhibitors of Pontin by combining
virtual screening and enzymatic assay
Judith ELKAIM*, Michel CASTROVIEJO†, Driss BENNANI*, Said TAOUJI‡, Nathalie ALLAIN‡, Michel LAGUERRE*,Jean ROSENBAUM‡, Jean and Patrick LESTIENNE*Molecular Modeling Group, IECB-CNRS-Universit´e de Bordeaux, UMR 5248, 2 rue R. Escarpit, F-33607 Pessac, France, †Platform Protein Expression and Purification, CNRS, UMR5234, 146 rue L. Saignat, F-33076 Bordeaux Cedex, France, and ‡Physiopathologie du Cancer du Foie, INSERM U1053-Universit´e de Bordeaux, 146 rue L. Saignat, F-33076 BordeauxCedex, France
functions coming from the same program were not consideredsince the functions are not independent
Individual scoring functions
The combinations of SURF with all functions from Xscore
DrugScore provides three scoring functions, i.e. PAIR, SURF
were explored first, since those functions were the best at
and PAIRSURF, and two alternative versions of DrugScore are
discriminating between the decoys individually. The consensuses
available: DrugScoreCSD and DrugScorePDB For both
obtained showed that the results given by those functions were
versions, SURF score is identical and PAIRSURF score is the sum
highly correlated, in particular in the consensus HPscore/SURF
of PAIR and SURF score. Consequently, five distinct functions
30, where up to 242 compounds out of 279 in the best 30 %
are available from DrugScore, that will be noted PAIR_csd,
were common to both top lists, including only one decoy. Similar
PAIRSURF_csd, PAIR_pdb, PAIRSURF_pdb and SURF. Xscore
results were observed with HSscore/SURF 30 with 217 common
offers four scoring functions that are HPscore, HMscore,
compounds, among which one was a decoy. HMscore/SURF 30
HSscore and AVEscore, with the last being the average of the
and AVEscore/SURF 30 gave poorer results, with 204 and 220
first three.
molecules respectively shared by both functions, including two
At first, all scoring functions were evaluated individually by
decoys. Yet, none of these consensuses was able to completely
comparing the number of decoys ranked in the best 15 % (139
exclude the decoys.
compounds) of the training set
The consensuses made with Vina displayed a much lower
All dockings were performed with Vina meaning that its
number of common ligands. In the consensus Vina/SURF 30, 127
scoring function was used during the process of poses generation.
molecules were shared by both functions, but two of these were
Therefore the correctness of this function was a key factor for
decoys. As for Vina/HMscore 30 and Vina/AVEscore 30, they
the accuracy of the whole experiment. Out of the 929 compounds
provided 128 and 121 shared compounds respectively, including
present in the training set, two decoys appeared in the top 15 %
one decoy. Vina/HSscore 30 and Vina/HPscore 30 did best at
with the scoring function inherent to Vina.
eliminating the decoys from the top since 119 and 125 compounds
The functions from DrugScoreCSD clearly failed to discriminate
respectively responded to both individual functions, with no
the decoys. With PAIRSURF_csd and PAIR_csd, five decoys were
found in the top 15 %. The PDB version of DrugScore performed
Using three functions, the consensuses Vina/HSscore/SURF
better, with both PAIRSURF_pdb and PAIR_pdb ranking only
30 and Vina/HPscore/SURF 30 gave very similar results, with
one decoy in the best 15 %. Similarly, SURF ranked only decoy
100 and 108 compounds respectively responding to the three
in the top 15 %.
functions, with no decoys.
The best results were obtained with the functions from Xscore.
AVEscore and HPscore both ranked only one decoy in the best15 %, and HSscore and HMscore were very efficient at excluding
Decoy discrimination, single functions
the decoys from the top, since no decoy was observed in the best
Number and ranks (in parentheses) of the decoys found in the top 15 % of the training set
(Calbiochem database + decoys).
From now on, a consensus will be noted as follows:
‘Function1/Function2 X', with X being the percentage of base
5 (21, 32, 57, 58, 81)
considered for each function. To discriminate the combinations
5 (30, 36, 43, 65, 104)
that were able to exclude the decoys from the top-scoring
compounds, the consensuses were first carried out by keeping up
to 30 % of the base for each function, i.e. 279 compounds, and then
progressively reducing the percentage of base considered. Using
the Consensus Scoring protocol from Discovery Studio 2.1, the
numerous combinations available with the ten scoring functions
were explored. However, the combinations of different scoring
1 Correspondence may be addressed to either of these authors (email [email protected] for molecular modelling aspects or
[email protected] for biochemical aspects).
� The Authors Journal compilation c
� 2012 Biochemical Society
J. Elkaim and others
Decoy discrimination, consensus
Number of common ligands in the consensus 30 % and number of decoys found among them(Calbiochem database + decoys).
1 Velec, H. F. G., Gohlke, H. and Klebe, G. (2005) DrugScoreCSD: knowledge-based scoring
function derived from small molecule crystal data with superior recognition rate of near
native ligand poses and better affinity prediction. J. Med. Chem. 48, 6296–6303
2 Gohlke, H., Hendlich, M. and Klebe, G. (2000) Knowledge-based scoring function to
predict protein–ligand interactions. J. Mol. Biol. 295, 337–356
3 Wang, R., Laib, L. and Wang, S. (2002) Further development and validation of empirical
scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol.
Des. 16, 11–26
4 Trott, O. and Olson, A. J. (2010) AutoDock Vina: Improving the speed and accuracy of
docking with a new scoring function, efficient optimization, and multithreading. J. Comput.
Chem. 31, 455–461
5 Wang, R. and Wang, S. (2001) How does consensus scoring work for virtual library
screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 41, 1422–1426
Received 5 October 2011/23 January 2012; accepted 24 January 2012Published as BJ Immediate Publication 24 January 2012, doi:10.1042/BJ20111779
� The Authors Journal compilation c
� 2012 Biochemical Society
Source: http://chimiotheque-nationale.cn.cnrs.fr/IMG/pdf/BiochemJ2012_1_.pdf
SAFETY PROFILE OF RUPATADINE IN THE TREATMENT OF CHRONIC URTICARIA Giménez-Arnau A1, Malbran A2, Poop G3, Benea V4, Medina I5, Garcia O6, Donado E6 1Hospital del Mar, Dermatology Department, IMAS. Barcelona, Spain. 2Unidad de Alergia, Asma o Inmunología-COM. Buenos Aires, Argentina. 3Dermatological Clinical Practica. Augsburg, Germany. 4Clinical Hospital Prof Scarlat Longhin. Bucharest, Romania.
PRODUCT MONOGRAPH Pr FAMPYRA™ 10 mg Sustained Release Tablet Potassium Channel Blocker Biogen Idec Canada Inc. Date of Revision: 90 Burnhamthorpe Road West, Suite 1100 November 26, 2014 Mississauga, Ontario L5B 3C3 Submission Control No: 177177 Page 1 of 32 TABLE OF CONTENTS PART I: HEALTH PROFESSIONAL INFORMATION.3 SUMMARY PRODUCT INFORMATION .3 INDICATIONS AND CLINICAL USE .3 CONTRAINDICATIONS .3 WARNINGS AND PRECAUTIONS .4 ADVERSE REACTIONS .9 DRUG INTERACTIONS .14 DOSAGE AND ADMINISTRATION .15 OVERDOSAGE .16 ACTION AND CLINICAL PHARMACOLOGY .17 STORAGE AND STABILITY .19 SPECIAL HANDLING INSTRUCTIONS .19 DOSAGE FORMS, COMPOSITION AND PACKAGING .19 PART II: SCIENTIFIC INFORMATION .20 PHARMACEUTICAL INFORMATION .20 CLINICAL TRIALS .21 DETAILED PHARMACOLOGY .24 TOXICOLOGY .25 REFERENCES .27 PART III: CONSUMER INFORMATION .28


