
Cialis ist bekannt für seine lange Wirkdauer von bis zu 36 Stunden. Dadurch unterscheidet es sich deutlich von Viagra. Viele Schweizer vergleichen daher Preise und schauen nach Angeboten unter dem Begriff cialis generika schweiz, da Generika erschwinglicher sind.
Microsoft word - acembl_manual_2009_1

ACEMBL Expression System
User Manual
Vers. 09.11
Yan Nie, Christoph Bieniossek, Imre Berger
ACEMBL was developed at the European Molecular Biology Laboratory EMBL Grenoble Outstation 38042 Grenoble CEDEX 9, France Grenoble, August 21, 2009
ACEMBL System User Manual
EMBL Grenoble, 2009
Table of Contents
A. Synopsis:
B. ACEMBL System
B.1. ACEMBL vectors
B.2. The multiple integration element (MIE)
B.3. Tags, promoters, terminators
B.4. Complex Expression
C. Procedures
C.1. Cloning into ACEMBL vectors
C1.1. Single gene insertion into the MIE by SLIC
C1.2. Polycistron assembly in MIE by SLIC
C.1.3. Gene insertion by restriction/ligation
C.1.4. Multiplication by using the HE and BstXI sites
C.2. Cre-LoxP reaction of Acceptors and Donors
C.2.1. Cre-LoxP fusion of Acceptors and Donors
C.2.2. Deconstruction of fusion vectors by Cre
C.3. Coexpression by Cotransformation
D. ACEMBL multigene combination: Examples
D.1. SLIC cloning into ACEMBL vectors: human TFIIF
D.2. Polycistron by SLIC: human VHL/ElonginB/ElonginC complex.
D.3. The Homing endonuclease/BstXI module: yeast RES complex
D.4. Coexpression by cotransformation: human NYB/NYC
D.5. Coexpression from Acceptor-Donor fusions
E. The ACEMBL System Kit
F. Appendix
F.1. DNA sequence of MIE
F.2. DNA sequences of ACEMBL vectors
F.2.6. pACKS tetrafusion (ACEMBL kit component)
Protocols
Protocol 1: Single gene insertion by SLIC
Protocol 2: Polycistron assembly by SLIC
Protocol 3: Restriction/ligation cloning into the MIE.
Protocol 4: Multiplication by using homing endonuclease/BstXI
ACEMBL System User Manual
EMBL Grenoble, 2009
Table I: Adaptor DNA sequences.
Illustrations:
ACEMBL system for multiprotein complex production
The multiple integration element, schematic view
Single gene insertion by SLIC
Generating a polycistron by SLIC
LoxP imperfect inverted repeat
Cre and De-Cre reaction pyramid
96well analysis of Cre assembly
Multifragment SLIC of pACE-VHLbc (tricistron)
The HE/BstXI multiplication module
ACEMBL System Kit: Generating single vectors from pACKS
96well microtiter analysis of pACKS De-Cre reaction
ACEMBL plasmid maps
ACEMBL System User Manual
EMBL Grenoble, 2009
ACEMBL is a 3rd generation multigene expression system for complex production in E. coli, created at the European Molecular Biology Laboratory EMBL, at Grenoble. ACEMBL can be applied both manually and also in an automated setup by using a liquid handling workstation. ACEMBL applies tandem recombination steps for rapidly assembling many genes into multigene expression cassettes. These can be single or polycistronic expression modules, or a combination of these elements. ACEMBL also offers the option to employ conventional approaches involving restriction enzymes and ligases if desired, which may be the methods of choice in laboratories not familiar with recombination approaches.
The following strategies for multigene assembly and expression are provided
for in the ACEMBL system and detailed in Sections B and C:
(1) Single gene insertions into vectors (recombination or restriction/ligation)
(2) Multigene assembly into a polycistron (recombination or restriction/ligation)
(3) Multigene assembly using homing endonucleases
(4) Multigene plasmid fusion by Cre-LoxP reaction
(5) Multigene expression by cotransformation
These strategies can be used individually or in conjunction, depending on the project and user.
In Section C, step-by-step protocols are provided for each of the methods for
multigene cassette assembly that can be applied in the ACEMBL system. Each procedure is illustrated by corresponding complex expression experiments in Section D of this Supplement.
DNA sequences of ACEMBL vectors are provided in the Appendix and can
be copied from there for further use.
Requests for ACEMBL system kit components can be addressed to Imre
Berger ([email protected]).
ACEMBL System User Manual
EMBL Grenoble, 2009
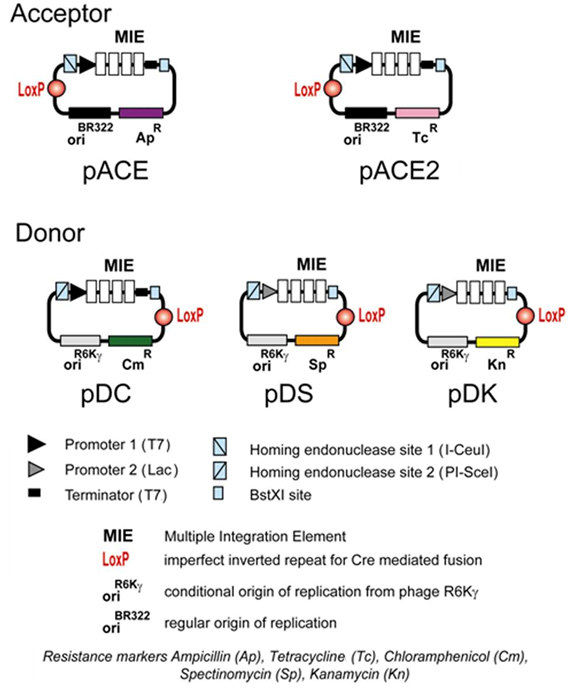
B. ACEMBL System B.1. ACEMBL vectors At the core of the technology are five small de novo designed vectors which are called "Acceptor" and "Donor" vectors (Illustration 1). Acceptor vectors (pACE, pACE2) contain origins of replication derived from ColE1 and resistance markers (ampicillin or tetracycline). Donor vectors contain conditional origins of replication (derived from R6Kγ), which make their propagation dependent on hosts expressing the pir gene. Donor vectors contain resistance markers kanamycin, chloramphenicol, spectinomycin. Up to three Donor vectors can be used in conjunction with one Acceptor vector
Illustration 1: ACEMBL system for multiprotein complex production.
All Donor and Acceptor vectors contain a loxP imperfect inverted repeat and
in addition, a multiple integration element (MIE). This MIE consists of an expression cassette with a promoter of choice (prokaryotic, mammalian, insect cell specific or a combination thereof) and a terminator (prokaryotic, mammalian, insect cell specific
ACEMBL System User Manual
EMBL Grenoble, 2009
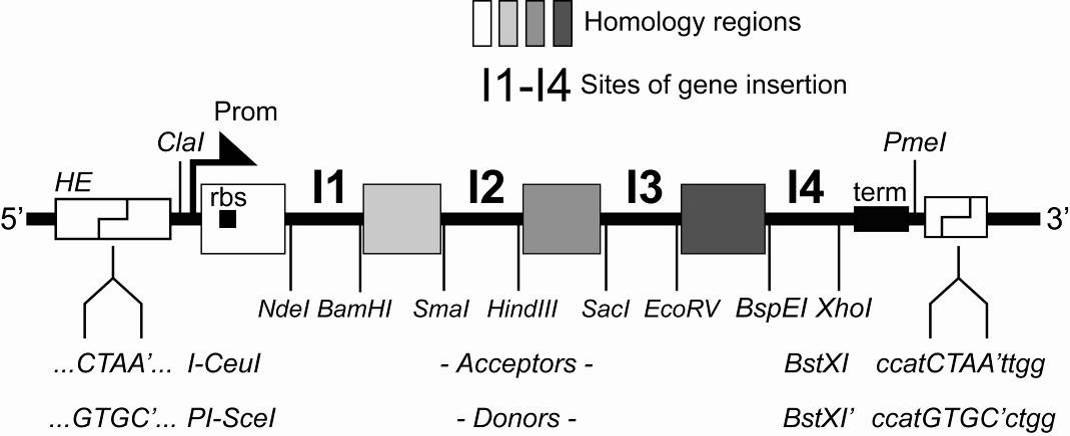
or a combination thereof). In between is a DNA segment which contains a number of restriction sites that can be used for conventional cloning approaches or also for generating double-strand breaks for the integration of expression elements of choice (further promoters, ribosomal binding sites, terminators and genes). The MIE is completed by a homing endonuclease site and a specifically designed restriction enzyme site (BstXI) flanking the promoter and the terminator (see B.2.) Vector DNA sequences are provided in the Appendix. Maps of all vectors are shown at the end of this manual. B.2. The multiple integration element (MIE)
Illustration 2: The multiple integration element, schematic view.
The MIE was derived from a polylinker1 and allows for several approaches for multigene assembly (Section C). Multiple genes can be inserted into the MIE of any one of the vectors by a variety of methods, for example BD-In-Fusion recombination2 or SLIC (sequence and ligation independent cloning3. For this, the vector needs to be linearized, which can also be carried out efficiently by PCR reaction with appropriate primers, since the vectors are all small (2-3.0 kb). Use of ultrahigh-fidelity polymerases such as Phusion4 is recommended. Alternatively, if more conventional approaches are preferred i.e. in a regular wet lab setting without robotics, the vectors can also be linearized by restriction digestion, and a gene of interest can be integrated by restriction / ligation (Section C). The DNA sequence of the MIE is shown in the Appendix.
1 Tan, S. et al. Protein Expr. Purif. 40, 385 (2005)
2 ClonTech TaKaRa Bio Europe, www.clontech.com
3 Li, M. and Elledge, S., Nat. Methods 4, 251 (2007)
4 Finnzymes/New England BioLabs, www.neb.com ACEMBL System User Manual
EMBL Grenoble, 2009
B.3. Tags, promoters, terminators Current vectors of the ACEMBL system contain per default promoters T7 and Lac, as well as the T7 terminator element (Illustr.1, 10). The T7 system is most commonly used currently; it requires bacterial strains which contain a T7 polymerase gene in the E. coli genome. The Lac promoter is a strong endogenous promoter which can be utilized in most strains. All ACEMBL vectors contain the lac operator element for repression of heterologous expression.
Evidently, all promoters and terminators present in ACEMBL Donor and
Acceptor vectors, and in fact the entire multiple integration element (MIE) can be exchanged with a favored expression cassette of choice by using restriction/ligation cloning with appropriate enzymes (for example ClaI/PmeI, Illustration 2) or insertion into linearized ACEMBL vectors where the MIE was removed by sequence and ligation independent approaches such as SLIC. We have substituted the T7 promoter in pDC with a trc promoter (pDCtrc), and the T7 promoter in pACE with an arabinose promoter (pACEara) and used the resulting vectors successfully in coexpression experiments by inducing with arabinose and IPTG.
Currently, the ACEMBL system vectors do not contain DNA sequences
encoding for affinity tags to facilitate purification or solubilization of the protein(s) of interest. We typically use C- or N-terminal oligohistidine tags, with or without protease sites for tag removal. We introduce these by means of the respective PCR primers used for amplification of the genes of interest prior to SLIC mediated insertion. We recommend to outfit Donors or Acceptors of choice by the array of custom tags that are favored in individual user laboratories prior to inserting recombinant genes of interest. This is best done by a design which will, after tag insertion, still be compatible with the recombination based principles of ACEMBL system usage.
ACEMBL System User Manual
EMBL Grenoble, 2009
B.4. Complex Expression
For expression in E.coli, the ACEMBL multigene expression vector fusions with appropriate promoters or terminators are transformed into the appropriate expression host of choice. In the current version (T7 and lac promoter elements), most of the wide array of currently available expression strains can be utilized. If particular expression strains already contain helper plasmids with DNA encoding for chaperones, lysozyme or else, the design of the multigene fusion should ideally be such that the ACEMBL vector containing the resistance marker that is also present on the helper plasmid is not included in multigene vector construction (although this is probably not essential).
Alternatively, the issue can be resolved by creating new versions of the
ACEMBL vectors containing resistance markers that circumvent the conflict. This can be easily performed by PCR amplifying the vectors minus the resistance marker, and combine the resulting fragments with a PCR amplified resistance marker by recombination (SLIC) or blunt-end ligation (using 5'phosphorylated primers). Note that resistance markers can also be exchanged in between ACEMBL vectors by restriction digestion with AlwNI and ClaI (for Donors) and AlwNI and PmeI (for Acceptors).
Donor vectors depend on the pir gene product expressed by the host, due to
the R6K conditional origin of replication. In regular expression strains, they rely on fusion with an Acceptor for productive replication. Donors or Donor-Donor fusions can nonetheless be used even for expression when not fused with an Acceptor, by using expression strains carrying a genomic insertion of the pir gene. Such strains have more recently become available (Novagen Inc., Madison WI, USA).
Cotransformation of two plasmids can also lead to successful protein complex
expression. The ACEMBL system contains two Acceptor vectors, pACE and pACE2, which are identical except for the resistance marker (Illustration 1). Therefore, genes present on pACE or pACE2, respectively, can be expressed by cotransformation of the two plasmids and subsequent exposure to both tetracyclin and ampicillin simultaneously. In fact, entire Acceptor-Donor fusions containing several genes, based on pACE or pACE2 as Acceptors, can in principle be cotransformed for mutli-expression, if needed.
ACEMBL System User Manual
EMBL Grenoble, 2009
C.1. Cloning into ACEMBL vectors
All Donors and Acceptors contain an identical MIE with exception of the homing endonuclease site / BstXI tandem encompassing the MIE (Illustrations 1 and 12). The MIE is tailored for sequence and ligation independent gene insertion methods. In addition, the MIE also contains a series of unique restriction sites, and therefore can be used as a classical polylinker for conventional gene insertion by restriction/ligation. We suggest to choose the methods a user lab is most proficient with. For automated applications, restriction/ligation is essentially ruled out. In this case, recombination approaches can be used efficiently for gene insertion (SLIC).
C1.1. Single gene insertion into the MIE by SLIC
Several procedures for restriction/ligation independent insertion of genes into vectors have been published or commercialized (Novagen LIC, Becton-Dickinson BD In-Fusion and others), each with its own merit. These systems share in common that they rely on the exonuclease activity of DNA polymerases. In the absence of dNTPs, 5' extensions are created from blunt ends or overhangs by digestion from the 3' end. If two DNA fragments contain the same 20 bp sequence at their termini at opposite ends, this results in overhangs that share complementary sequences capable of annealing. This can be exploited for ligation independent combination of two or several DNA fragments containing homologous sequences.
If T4 DNA polymerase is used, this can be carried out in a manner that is
independent of the sequences of the homology regions (Sequence and Ligation Independent Cloning, SLIC) and detailed protocols became available. In the context of multiprotein expression, this is particularly useful, as the presence of unique restriction sites, or their creation by mutagenesis, in the ensemble of encoding DNAs ceases to be an issue.
We adapted SLIC for inserting encoding DNAs amplified by Phusion
polymerase into the ACEMBL Acceptor and Donor vectors according to the published protocols. In this way, not only seamless integration of genes into the expression cassettes, but also concatamerization of expression cassettes to multigene constructs can be achieved by applying the same, simple routine that can be readily automated.
ACEMBL System User Manual
EMBL Grenoble, 2009
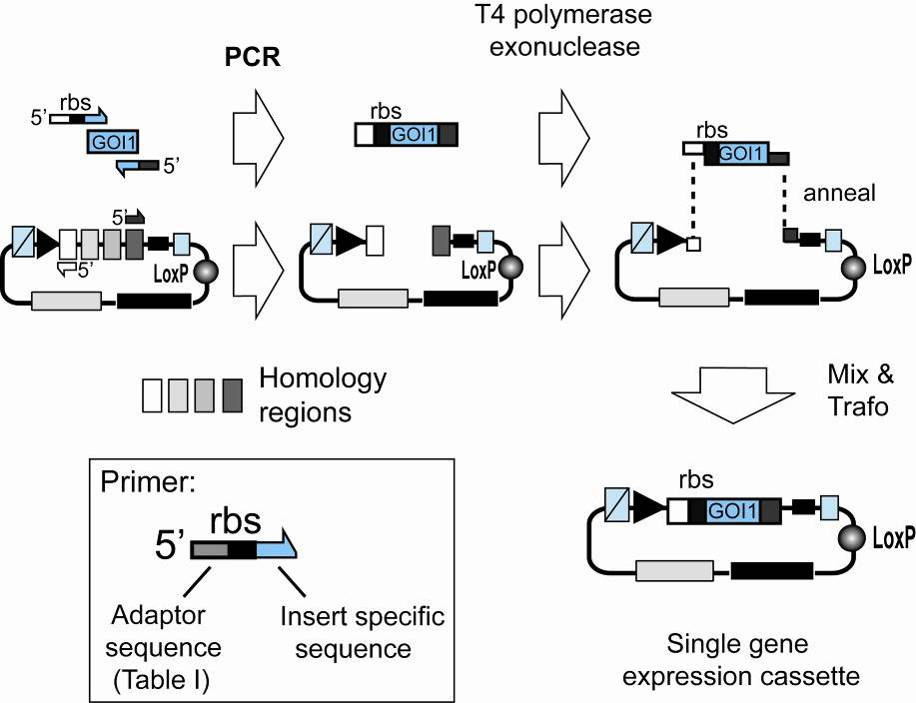
Illustration 3: Single gene insertion by SLIC. A gene of interest (GOI 1) is PCR
amplified with specific primers and integrated into a vector (Acceptor, Donor)
linearized by PCR with complementary primers (complementary regions are
shaded in light gray or dark grey, respectively). Resulting PCR fragments contain
homology regions at the ends. T4 DNA polymerase acts as an exonuclease in the
absence of dNTP and produces long sticky overhangs. Mixing (optionally
annealing) of T4DNA polymerase exonuclease treated insert and vector is
followed by transformation, yielding a single gene expression cassette.
We use an improved protocol for SLIC which was modified from the original
publication5. This protocol as applied manually is detailed below (Protocol 1). If other systems are used (BD-InFusion etc.), follow manufacturers' recommendations. For robotics applications, modifications of the protocol may be necessary and will be detailed elsewhere6.
Protocol 1: Single gene insertion by SLIC.
Reagents required:
Phusion Polymerase 5x HF Buffer for Phusion Polymerase dNTP mix (10 mM) T4 DNA polymerase (and10x Buffer) DpnI enzyme E. coli competent cells 100mM DTT, 2M Urea, 500 mM EDTA
5 Li, M. and Elledge, S., Nat. Methods 4, 251 (2007)
6 Bieniossek, C., Nie, Y. et al., in preparation.
ACEMBL System User Manual
EMBL Grenoble, 2009
Step 1: Primer design
Primers for the SLIC procedure are designed to provide the regions of homology which result in the long sticky ends upon treatment with T4 DNA polymerase in the absence of dNTP: Primers for the insert contain a DNA sequence corresponding to this region of homology (" Adaptor sequence" in Illustration 3, inset), followed by sequence which specifically anneals to the insert to be amplified Illustration 3, inset). Useful adaptor sequences for SLIC are listed below (Table I). If the gene of interest (GOI) is amplified from a vector already containing expression elements (e.g. the pET vector series), this " insert specific sequence" can be located upstream of a ribosome binding site (rbs). Otherwise, the forward primer needs to be designed such that a ribosome binding site is also provided in the final construct (Illustration 3, inset). Primers for PCR linearization of the vector backbone are simply complementary to the two adaptor sequences present in the primer pair chosen for insert amplification (Illustration 3).
Step 2: PCR amplification of insert and vector
Identical reactions are prepared in 100-µl volume for DNA insert to be cloned
and vector to be linearized by PCR:
5× Phusion HF Reaction buffer
dNTPs (10 mM stock)
Template DNA (100 ng/µl)
5′ SLIC primer (100 µM stock)
3′ SLIC primer (100 µM stock)
Phusion polymerase (2 U/µl)
PCR reactions are then carried out with a standard PCR program (unless very
long DNAs are amplified, then double extension time):
1 x 98° C for 2 min 30 x [98° C for 20 sec. -> 50°C for 30 sec. -> 72°C for 3 min] Hold at 10°C
Analysis of the PCR reactions by agarose gel electrophoresis and ethidium
bromide staining is recommended.
ACEMBL System User Manual
EMBL Grenoble, 2009
Step 3: DpnI treatment of PCR products (optional)
PCR reactions are then supplied with 1 µl DpnI enzyme which cleaves parental
plasmids (that are methylated). For insert PCR reactions, DpnI treatment is not
required if the resistance marker of the template plasmid differs from the
destination vector. Reactions are then carried out as follows:
Incubation: 37°C for 1-4h Inactivation: 80°C for 20 min
Step 4: Purification of PCR products
! PCR products must be cleaned of residual dNTPs !
Otherwise, the T4 DNA polymerase reaction (Step 5) is compromised. Product purification is best performed by using commercial PCR Purification Kits
or NulceoSpin Kits (Qiagen, MacheryNagel or others). It is recommended to
perform elution in the minimal possible volume indicated by the manufacturer.
Step 5: T4 DNA polymerase exonuclease treatment
Identical reactions are prepared in 20-µl volume for insert and for vector (eluted
10x T4 DNA polymerase buffer
DNA eluate from Step 3 (vector or insert) 14 µl T4 DNA polymerase
Reactions are then carried out as follows:
Incubation: 23°C for 20 min Arrest:
Addition of 1 µl 500 mM EDTA
Inactivation: 75°C for 20 min
Step 6: Mixing and Annealing
T4 DNA polymerase exonuclease treated insert and vector are then mixed,
followed by an (optional) annealing step which was found to enhance efficiency7:
T4 DNA pol treated insert:
T4 DNA pol treated vector:
Annealing: 65°C for 10 min
Slowly (in heat block) to RT
Step 7: Transformation
Mixtures are next transformed into competent cells following standard
transformation procedures. Reactions for pACE and pACE2 derivatives are transformed into standard E. coli cells for cloning (such as TOP10, DH5α, HB101) and after recovery (204h)
7 Dr. Rolf Jaussi, PSI Villigen, personal communication
ACEMBL System User Manual
EMBL Grenoble, 2009
plated on agar containing ampicillin (100 µg/ml) or tetracycline (25 µg/ml),
respectively. Reactions for Donor derivatives are transformed into E. coli cells expressing the
pir gene (such as BW23473, BW23474, or PIR1 and PIR2, Invitrogen) and plated on agar containing chloramphenicol (25 µg/ml, pDC), kanamycin (50 µg/ml,
pDK), and spectinomycin (50 µg/ml, pDS).
Step 8: Plasmid analysis
Plasmids are cultured in small-scale in media containing the corresponding
antibiotic, and analyzed by sequencing and (optionally) restriction mapping with
an appropriate restriction enzyme.
C1.2. Polycistron assembly in MIE by SLIC
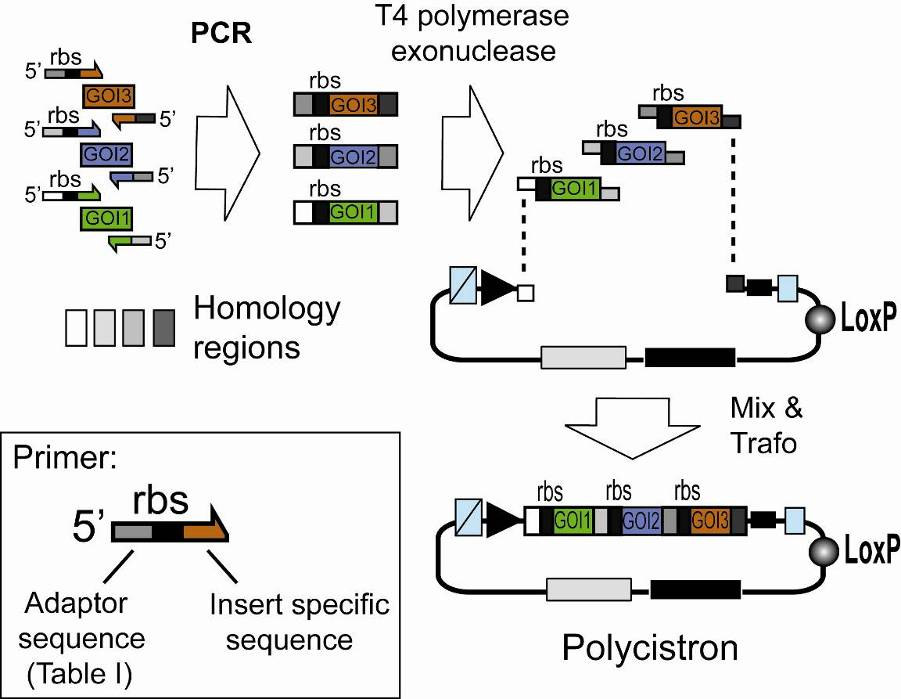
The multiple integration element can also be used to integrate genes of interest by using multi-fragment SLIC recombination as shown in Illustration 4. Genes preceded by ribosome binding sites (rbs) can be assembled in this way into polycistrons.
Illustration 4: Generating a polycistron by SLIC. Genes of interest (GOI
1,2,3) are PCR amplified with specific primers and integrated into a vector (Acceptor, Donor) linearized by PCR with primers complementary to the ends of the forward primer of the first (GOI 1) and the reverse primer of the last (GOI 3) gene to be assembled in the polycistron (complementary regions are shaded in light gray or dark grey, respectively). Resulting PCR fragments contain homology regions at the ends. T4 DNA polymerase acts as an exonuclease in the absence of dNTP and produces long sticky overhangs. Mixing (optionally annealing) of T4DNA polymerase exonuclease treated insert and vector is followed by transformation, yielding a polycistronic expression cassette.
ACEMBL System User Manual
EMBL Grenoble, 2009
Protocol 2. Polycistron assembly by SLIC.
Reagents required:
Phusion Polymerase 5x HF Buffer for Phusion Polymerase dNTP mix (10 mM) T4 DNA polymerase (and 10x Buffer) E. coli competent cells 100mM DTT, 2M Urea, 500 mM EDTA
Step 1: Primer design
The MIE element is composed of tried-and-tested primer sequences. These constitute the " Adaptor" sequences that can be used for inserting single genes or multigene constructs. Recommended adaptor sequences are listed below (Table I). Adaptor sequences form the 5' segments of the primers used to amplify DNA fragments to be inserted into the MIE. Insert specific sequences are added at 3', DNE encoding for a ribosome binding sites can be inserted optionally if not already present on the PCR template
Step 2: PCR amplification of insert and primer
Identical reactions are prepared in 100-µl volume for all DNA insert (GOI 1,2,3)
to be cloned and the vector to be linearized by PCR:
5× Phusion HF Reaction buffer
dNTPs (10 mM stock)
Template DNA (100 ng/µl)
5′ SLIC primer (100 µM stock)
3′ SLIC primer (100 µM stock)
Phusion polymerase (2 U/µl)
PCR reactions are then carried out with a standard PCR program (unless very
long DNAs are amplified, then double extension time):
1 x 98° C for 2 min 30 x [98° C for 20 sec. -> 50°C for 30 sec. -> 72°C for 3 min] Hold at 10°C
ACEMBL System User Manual
EMBL Grenoble, 2009
Analysis of the PCR reactions by agarose gel electrophoresis and ethidium
bromide staining is recommended.
Step 3: DpnI treatment of PCR products (optional)
PCR reactions are then supplied with 1 µl DpnI enzyme which cleaves parental
plasmids (that are methylated). For insert PCR reactions, DpnI treatment is not
required if the resistance marker of the template plasmids differs from the
destination vector. Reactions are then carried out as follows:
Incubation: 37°C for 1-4h Inactivation: 80°C for 20 min
Step 4: Purification of PCR products
! PCR products must be cleaned of residual dNTPs !
Otherwise, the T4 DNA polymerase reaction (Step 5) is compromised.
Product purification is best performed by using commercial PCR Purification Kits
or NulceoSpin Kits (Qiagen, MacheryNagel or others). It is recommended to
perform elution in the minimal possible volume indicated by the manufacturer.
Step 5: T4 DNA polymerase exonuclease treatment
Identical reactions are prepared in 20-µl volume for each insert (GOI 1,2,3) and
for the vector (eluted in Step 4):
10x T4 DNA polymerase buffer
DNA eluate from Step 3 (vector or insert) 14 µl T4 DNA polymerase
Reactions are then carried out as follows:
Incubation: 23°C for 20 min Arrest:
Addition of 1 µl 500 mM EDTA
Inactivation: 75°C for 20 min
Step 6: Mixing and Annealing
T4 DNA polymerase exonuclease treated insert and vector are then mixed,
followed by an (optional) annealing step which was found to enhance efficiency8:
T4 DNA pol treated insert 1 (GOI 1):
T4 DNA pol treated insert 2 (GOI 2):
T4 DNA pol treated insert 3 (GOI 3):
T4 DNA pol treated vector:
Annealing: 65°C for 10 min
Slowly (in heat block) to RT
8 Dr. Rolf Jaussi, PSI Villigen, personal communication . ACEMBL System User Manual
EMBL Grenoble, 2009
Step 7: Transformation
Mixtures are next transformed into competent cells following standard
transformation procedures. Reactions for pACE and pACE2 derivatives are transformed into standard E. coli cells for cloning (such as TOP10, DH5α, HB101) and after recovery plated on agar containing ampicillin (100 µg/ml) or tetracycline (25 µg/ml), respectively. Reactions for Donor derivatives are transformed into E. coli cells expressing the
pir gene (such as BW23473, BW23474, or PIR1 and PIR2, Invitrogen) and plated on agar containing chloramphenicol (25 µg/ml, pDC), kanamycin (50 µg/ml,
pDK), and spectinomycin (50 µg/ml, pDS).
Step 8: Plasmid analysis
Plasmids are cultured and correct clones are selected based on specific restriction
digestion and DNA sequencing of the inserts.
Table I. Adaptor sequences ! updated to commercially available kit, ATG-biosynthetics !
For single gene or multigene insertions into ACEMBL vectors by SLIC.
Adaptor1
Sequence
Description
Forward primer for insert amplification, if gene of
interest (GOI) is present in a T7 system vector (i.e.
pET series). No further extension (rbs, insert specific overlap)
Reverse primer for insert amplification, if GOI is
CCCCAAGGGGTTATGCTAG
present in a T7 system vector (i.e. pET series). No further extension (stop codon, insert specific
overlap) required.
Forward primer for vector amplification, reverse
CTCTAAACGGGTCTTGAGG
complement of T7InsRev. No further extension required.
Reverse primer for vector amplification, reverse
complement of T7InsFor. No further extension required.
Forward primer for insert amplification for
insertion into MIE site I1 (Illustration 2). Further extension at 3' (insert specific overlap)
required. Can be used with adaptor XhoInsRev in case of
single fragment SLIC (Illustr. 3).
pACE,pACE2,pDC (T7):
Reverse primer for insert amplification for
insertion into MIE site I4 (Illustr. 2).
Further extension at 3' (stop codon, insert specific
overlap) required.
Can be used with adaptor NdeInsFor in case of
single fragment SLIC (Illustr. 3).
ACEMBL System User Manual
EMBL Grenoble, 2009
pACE,pACE2,pDC (T7):
Forward primer for vector amplification, reverse
complement of XhoInsRev
No further extension required.
pDK,pDS (lac): CTCGAGACTAGTTCCGTTTAAACCC
Reverse primer for vector amplification, reverse
complement of NdeInsFor. No further extension required.
Reverse primer for insert amplification (GOI1) for
insertion into MIE site I1 (Illustr. 2). Further extension at 3' (stop codon, insert specific
overlap) required. Use with adaptor NdeInsFor.
Forward primer for insert amplification (GOI2) for
insertion into site I2 (Illustr. 2,4). Further extension at 3' (rbs, insert specific
overlap) required. Use with adaptor SacHind (multifragment SLIC,
Reverse primer for insert amplification (GOI2)
insertion into MIE site I2 (Illustr. 2, 4). Further extension at 3' (stop codon, insert specific
overlap) required. Use with adaptor BamSma (multifragment SLIC,
Forward primer for insert amplification (GOI3) for
insertion into site I3 (Illustr. 2,4). Further extension at 3' (rbs, insert specific
overlap) required. Use with adaptor BspEco (multifragment SLIC,
Reverse primer for insert amplification (GOI3)
insertion into MIE site I3 (Illustr. 2, 4). Further extension at 3' (stop codon, insert specific
overlap) required. Use with adaptor HindSac.(multifragment SLIC,
Forward primer for insert amplification (GOI4) for
insertion into site I4 (Illustr. 2,4). Further extension at 3' (rbs, insert specific
overlap) required. Use with adaptor XhoInsRev (multifragment
SLIC, Illustr. 4)
1 All Adaptor primers (without extension) can be used as sequencing primers for
genes of interest that were inserted into the MIE.
ACEMBL System User Manual
EMBL Grenoble, 2009
C.1.3. Gene insertion by restriction/ligation
The MIE can also be interpreted as a simple multiple cloning site with a series of unique restriction sites. The MIE is preceded by a promoter and a ribosome binding site, and followed by a terminator, therefore, cloning into the MIE by classical restriction/ligation also yields functional expression cassettes.
Genes of interest can be subcloned by using standard cloning procedures into
the multiple integration element (MIE) (see Appendix) of ACEMBL vectors (the MIE is identical in all vectors).
Protocol 3. Restriction/ligation cloning into the MIE.
Reagents required:
Phusion Polymerase 5x HF Buffer for Phusion Polymerase dNTP mix (10 mM) 10 mM BSA Restriction endonucleases (and 10x Buffer) T4 DNA ligase (and 10x Buffer) Calf or Shrimp intestinal alkaline phosphatase E. coli competent cells
Step 1: Primer design
For conventional cloning, PCR primers are designed containing chosen restriction
sites, preceded by appropriate overhangs for efficient cutting (c.f. New England
Biolabs catalogue), and followed by 20 nucleotides overlapping with the gene of
interest that is to be inserted. All MIEs are identical in the ACEMBL vectors. They contain a ribosome binding
preceding the NdeI site. For single gene insertions, therefore, a rbs need not be
included in the primer. If multigene insertions are planned (for example in insertion sites I1-I4 of the
MIE), primers need to be designed such that a rbs preceding the gene and a stop
codon at its end are provided. In particular for polycistron cloning by restriction/ligation, is recommended to
construct templates by custom gene synthesis. In the process, the restriction sites
present in the MIE can be eliminated from the encoding DNAs.
ACEMBL System User Manual
EMBL Grenoble, 2009
Step 2: Insert preparation
PCR of insert(s): Identical PCR reactions are prepared in 100 µl volume for genes of interest to be
inserted into the MIE:
5× Phusion HF Reaction buffer
dNTPs (10 mM stock)
Template DNA (100 ng/µl)
5′ primer (100 µM stock)
3′ primer (100 µM stock)
Phusion polymerase (2 U/µl)
PCR reactions are then carried out with a standard PCR program (unless very
long DNAs are amplified, then double extension time):
1 x 98° C for 2 min 30 x [98° C for 20 sec. -> 50°C for 30 sec. -> 72°C for 3 min] Hold at 10°C
Analysis of the PCR reactions by agarose gel electrophoresis and ethidium
bromide staining is recommended.
Product purification is best performed by using commercial PCR Purification
Kits or NulceoSpin Kits (Qiagen, MacheryNagel or others). It is recommended to
perform elution in the minimal possible volume indicated by the manufacturer.
Restriction digestion of insert(s): Restriction reactions are carried out in 40 µl reaction volumes, using specific
restriction enzymes as specified by manufacturer's recommendations (c.f. New
England Biolabs catalogue and others).
PCR Kit eluate ( 1 µg)
10x Restriction enzyme buffer
Restriction enzyme for 5'
Restriction enzyme for 3'
2 µl (in case of double
digestion, otherwise
Restriction digestions are performed in a single reaction with both enzymes
(double digestion) or sequentially (two single digestions) if the buffer conditions
required are incompatible.
Gel extraction of insert(s): Processed insert is then purified by agarose gel extraction using commercial kits
(Qiagen, MachereyNagel etc). It is recommended to elute the extracted DNA in
the minimal volume defined by the manufacturer.
ACEMBL System User Manual
EMBL Grenoble, 2009
Step 3: Vector preparation
Restriction digestion of ACEMBL plasmid(s): Restriction reactions are carried out in 40 µl reaction volumes, using specific
restriction enzymes as specified by manufacturer's recommendations (c.f. New
England Biolabs catalogue and others).
ACEMBL plasmid ( 0.5 µg) in ddH2O
10x Restriction enzyme buffer
Restriction enzyme for 5'
Restriction enzyme for 3'
2 µl (in case of double
digestion, otherwise
Restriction digestions are performed in a single reaction with both enzymes
(double digestion) or sequentially (two single digestions) if the buffer conditions
required are incompatible. Gel extraction of vector(s):
Processed vector is then purified by agarose gel extraction using commercial kits
(Qiagen, MachereyNagel etc). It is recommended to elute the extracted DNA in
the minimal volume defined by the manufacturer.
Step 4: Ligation
Ligation reactions are carried out in 20 µl reaction volumes according to the
recommendations of the supplier of T4 DNA ligase:
ACEMBL plasmid (gel extracted)
Insert (gel extracted)
10x T4 DNA Ligase buffer
Ligation reactions are performed at 25ºC (sticky end) for 1h or at 16ºC (blunt
Step 5: Transformation
Mixtures are next transformed into competent cells following standard
transformation procedures. Reactions for pACE and pACE2 derivatives are transformed into standard E. coli cells for cloning (such as TOP10, DH5α, HB101) and after recovery plated on agar containing ampicillin (100 µg/ml) or tetracycline (25 µg/ml), respectively. Reactions for Donor derivatives are transformed into E. coli cells expressing the
pir gene (such as BW23473, BW23474, or PIR1 and PIR2, Invitrogen) and plated on agar containing chloramphenicol (25 µg/ml, pDC), kanamycin (50 µg/ml,
pDK), and spectinomycin (50 µg/ml, pDS).
Step 6: Plasmid analysis
Plasmids are cultured and correct clones are selected based on specific restriction
digestion and DNA sequencing of the inserts.
ACEMBL System User Manual
EMBL Grenoble, 2009
C.1.4. Multiplication by using the HE and BstXI sites
All ACEMBL system vectors contain a homing endonuclease (HE) site and a designed BstXI site that envelop the multiple integration element (MIE). The homing endonuclease site can be used to insert entire expression cassettes, containing single genes or polycistrons, into a vector already containing one gene or several genes of interest. Homing endonucleases have long recognition sites (20-30 base pairs or more). Although not all equally stringent, homing endonuclease sites are most probably unique in the context of even large plasmids, or, in fact, entire genomes.
In the ACEMBL system, Donor vectors contain a recognition site for homing
endonuclease PI-SceI (Illustr. 2). This HE site yields upon cleavage a 3' overhang with the sequence -GTGC. Acceptor vectors contain the homing endonuclease site I-CeuI, which upon cleavage will result in a 3' overhang of -CTAA. On Acceptors and Donors, the respective HE site is preceding the MIE. The 3' end of the MIE contains a specifically designed BstXI site, which upon cleavage will generate a matching overhang. The basis of this is the specificity of cleavage by BstXI. The recognition sequence of BstXI is defined as CCANNNNN'NTGG (apostrophe marks position of phosphodiester link cleavage). The residues denoted as N can be chosen freely. Donor vectors thus contain a BstXI recognition site of the sequence CCATGTGC'CTGG, and Acceptor vectors contain CCATCTAA'TTGG. The overhangs generated by BstXI cleavage in each case will match the overhangs generated by HE cleavage. Note that Acceptors and Donors have different HE sites.
The recognition sites are not symmetric. Therefore, ligation of a HE/BstXI
digested fragment into a HE site of an ACEMBL vector will be (1) directional and (2) result in a hybrid DNA sequence where a HE halfsite is combined with a BstXI halfsite. This site will be cut by neither HE nor BstXI. Therefore, in a construct that had been digested with a HE, insertion by ligation of HE/BstXI digested DNA fragment containing an expression cassette with one or several genes will result in a construct which contains all heterologous genes of interest, enveloped by an intact HE site in front, and a BstXI site at the end. Therefore, the process of integrating entire expression cassettes by means of HE/BstXI digestion and ligation into a HE site can be repeated iteratively.
ACEMBL System User Manual
EMBL Grenoble, 2009
Protocol 4. Multiplication by using homing endonuclease/BstXI.
Reagents required:
Homing endonucleases PI-SceI, I-CeuI 10x Buffers for homing endonucleases Restriction enzyme BstXI (and 10x Buffer) T4 DNA ligase (and 10x Buffer) E. coli competent cells
Step 2: Insert preparation
Restriction reactions are carried out in 40 µl reaction volumes, using homing
endonucleases PI-SceI (Donors) or I-CeuI (Acceptors) as recommended by the
supplier (c.f. New England Biolabs catalogue and others).
ACEMBL plasmid ( 0.5 µg) in ddH2O 32 µl 10x Restriction enzyme buffer
PI-SceI (Donors) or I-CeuI (acceptors)
Reactions are then purified by PCR extraction kit or acidic ethanol precipitation,
and next digested by BstXI according to the recommendations of the supplier.
HE digested DNA in ddH2O
10x Restriction enzyme buffer
Gel extraction of insert(s): Processed insert is then purified by agarose gel extraction using commercial kits
(Qiagen, MachereyNagel etc). It is recommended to elute the extracted DNA in
the minimal volume defined by the manufacturer.
Step 3: Vector preparation
Restriction reactions are carried out in 40 µl reaction volumes, using homing
endonucleases PI-SceI (Donors) or I-CeuI (Acceptors) as recommended by the
supplier (c.f. New England Biolabs catalogue and others).
ACEMBL plasmid ( 0.5 µg) in ddH2O 33 µl 10x Restriction enzyme buffer
PI-SceI (Donors) or I-CeuI (acceptors)
ACEMBL System User Manual
EMBL Grenoble, 2009
Reactions are then purified by PCR extraction kit or acidic ethanol precipitation,
and next treated with intestinal alkaline phosphatase according to the
recommendations of the supplier.
HE digested DNA in ddH2O
10x Alkaline phosphatase buffer
Alkaline phosphatase
Gel extraction of vector: Processed vector is then purified by agarose gel extraction using commercial kits
(Qiagen, MachereyNagel etc). It is recommended to elute the extracted DNA in
the minimal volume defined by the manufacturer.
Step 4: Ligation
Ligation reactions are carried out in 20 µl reaction volumes:
HE/Phosphatase treated vector (gel extracted) 4 µl HE/BstXI treated insert (gel extracted)
10x T4 DNA Ligase buffer
Ligation reactions are performed at 25ºC for 1h or at 16ºC overnight.
Step 5: Transformation
Mixtures are next transformed into competent cells following standard
transformation procedures. Reactions for pACE and pACE2 derivatives are transformed into standard E. coli cells for cloning (such as TOP10, DH5α, HB101) and after recovery plated on agar containing ampicillin (100 µg/ml) or tetracycline (25 µg/ml), respectively. Reactions for Donor derivatives are transformed into E. coli cells expressing the
pir gene (such as BW23473, BW23474, or PIR1 and PIR2, Invitrogen) and plated on agar containing chloramphenicol (25 µg/ml, pDC), kanamycin (50 µg/ml,
pDK), and spectinomycin (50 µg/ml, pDS).
Step 6: Plasmid analysis
Plasmids are cultured and correct clones selected based on specific restriction
digestion and DNA sequencing of the inserts.
Note: Integration can likewise be performed by sequence and ligation independent
cloning. It is recommended to carry out linearization of the vector by digestion with
HE, if heterologous genes are already present, to avoid PCR amplifications over
encoding regions. The fragment to be inserted is generated by PCR amplification
resulting in a PCR fragment containing a 20-25 base pair stretch at its 5' end that is
identical to the corresponding DNA sequence present at the HE site counted from the
site of cleavage towards 5' (site of cleavage is position -4). At the 3' end of the PCR
fragment, the homology region is 20-25 base pairs counted from the site of cleavage
towards 3'.
ACEMBL System User Manual
EMBL Grenoble, 2009
C.2. Cre-LoxP reaction of Acceptors and Donors Cre recombinase is a member of the integrase family (Type I topoisomerase from bacteriophage P1). It recombines a 34 bp loxP site in the absence of accessory protein or auxiliary DNA sequence. The loxP site is comprised of two 13 bp recombinase-binding elements arranged as inverted repeats which flank an 8 bp central region where cleavage and ligation reaction occur.
The site-specific recombination mediated by Cre recombinase involves the
formation of a Holliday junction (HJ). The recombination events catalyzed by Cre
recombinase are dependent on the location and relative orientation of the loxP sites.
Two DNA molecules, for example an Acceptor and a Donor plasmid, containing
single loxP sites will be fused. Furthermore, the Cre recombination is an equilibrium
reaction with 20-30% efficiency in recombination. This provides useful options for
multigene combinations for multiprotein complex expression.
Illustration 5: LoxP imperfect inverted repeat
5'…ATAACTTCGTATA GCATACAT TATACGAAGTTAT…3' 3'…TATTGAAGCATAT CGTATGTA ATATGCTTCAATA…5'
inverted repeat
inverted repeat
In a reaction where several DNA molecules such as Donors and Acceptors are
incubated with Cre recombinase, the fusion/excision activity of the enzyme will result in an equilibrium state where single vectors (educt vectors) and all possible fusions coexist. Donor vectors can be used with Acceptors and/or Donors, likewise for Acceptor vectors. Higher order fusions are also generated where more than two vectors are fused. This is shown schematically in Illustration 6.
The fact that Donors contain a conditional origin of replication that depends
on a pir+ (pir positive) background now allows for selecting out from this reaction mix all desired Acceptor-Donor(s) combinations. For this, the reaction mix is used to transform to pir negative strains (TOP10, DH5α, HB101 or other common laboratory cloning strains). Then, Donor vectors will act as suicide vectors when plated out on agar containing the antibiotic corresponding to the Donor encoded resistance marker, unless fused with an Acceptor. By using agar with the appropriate combinations of antibiotics, all desired Acceptor-Donor fusions can be selected for.
ACEMBL System User Manual
EMBL Grenoble, 2009
We have generated fusion vectors of 25 kb and larger. In stability tests (serial
passaging for more than 60 generations), even such large plasmids proved to be stable as checked by restriction mapping, even if only one of the antibiotics corresponding to the encoded resistance markers was provided in the growth medium.
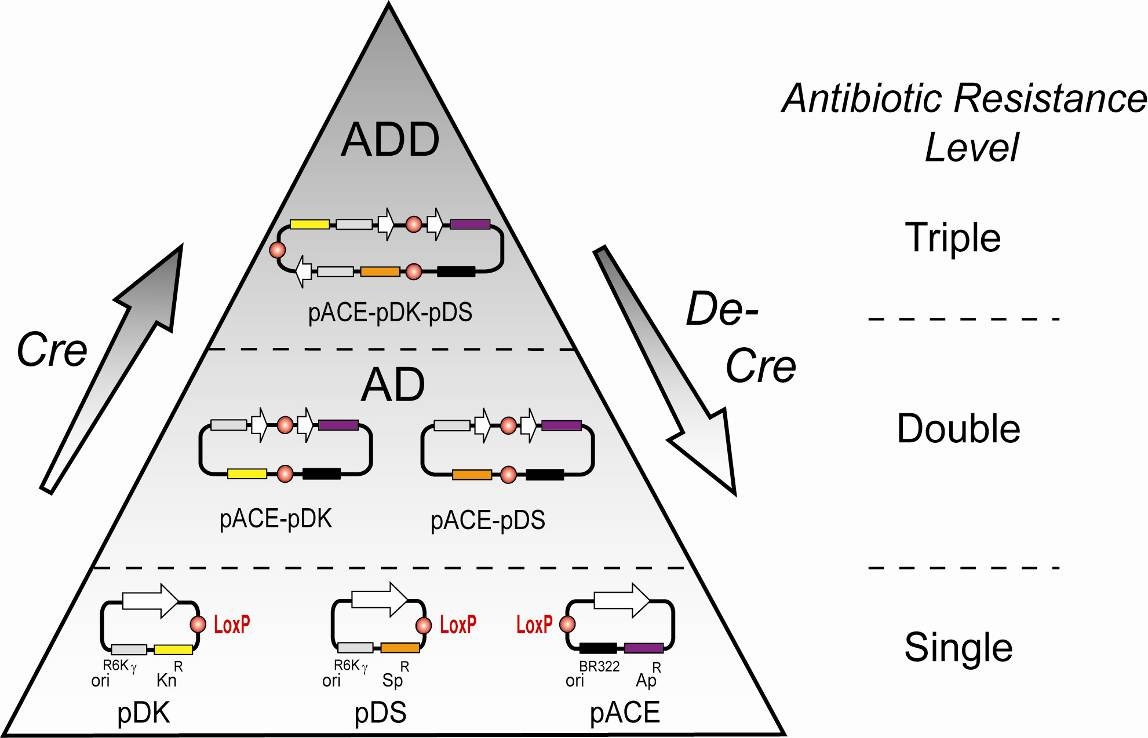
Illustration 6: Cre and De-Cre reaction pyramid
Cre-mediated assembly and disassembly of pACE, pDK, and pDS vectors are
shown in a schematic representation (left). LoxP sites are shown as red
circles, resistance markers and origins are labelled. White arrows stand for the
entire expression cassette (including promoter, terminator and multiple
integration elements) in the ACEMBL vectors. Not all possible fusion
products are shown for clarity. Levels of multiresistance are indicated (right).
C.2.1. Cre-LoxP fusion of Acceptors and Donors
This protocol is designed for generating multigene fusions from Donors and Acceptors by Cre-LoxP reaction.
Cre recombinase (from NEB or self made) Standard E. coli competent cells (pir- strain) Antibiotics 96well microtiter plates 12 well tissue-culture plates (or petri dishes) w. agar/antibiotics LB media
ACEMBL System User Manual
EMBL Grenoble, 2009
1. For a 20µl Cre reaction, mix 1 2 µg of each educt in approximately equal
amounts. Add ddH2O to adjust the total volume to 16 17 µl, then add 2 µl
10x Cre buffer and 1 2µl Cre recombinase.
2. Incubate Cre reaction at 37°C (or 30°C) for 1 hour.
3. Optional: load 2-5 µl of Cre reaction on an analytical agarose gel for
examination. Heat inactivation at 70°C for 10 minutes before the gel loading is strongly
recommended.
4. For chemical transformation, mix 10-15µl Cre reaction with 200 µl chemical
competent cells. Incubate the mixture on ice for 15-30 minutes. Then perform
heat shock at 42°C for 45-60 s. Up to 20 µl Cre reaction (0.1 volumes of the chemical competent cell suspension)
can be directly transformed into 200 µl chemical competent cells.
For electrotransformation, up to 2 µl Cre reaction could be directly mixed
with 100 µl electrocompetent cells, and transformed by using an
electroporator (e.g. BIORAD E. coli Pulser) at 1.8-2.0 kV. Larger volume of Cre reaction must be desalted by ethanol precipitation or PCR
purification column before electrotransformation. The desalted Cre reaction mix
should not exceed 0.1 volumes of the electrocompetent cell suspension. The cell/DNA mixture could be immediately used for electrotransformation without
prolonged incubation on ice.
5. Add up to 400 µl of LB media (or SOC media) per 100 µl of cell/DNA
suspension immediately after the transformation (heat shock or
electroporation).
6. Incubate the suspension in a 37°C shaking incubator overnight or for at least 4
hours (recovery period). For recovering multifusion plasmid containing more than 2 resistance markers, it is
strongly recommended to incubate the suspension at 37°C overnight.
7. Plate out the recovered cell suspension on agar containing the desired
combination of antibiotics. Incubate at 37°C overnight.
8. Clones from colonies present after overnight incubation can be verified by
restriction digestion at this stage (refer to steps 12-16). Especially in the case that only one multifusion plasmid is desired.
For further selection by single antibiotic challenges on a 96 well microtiter
plate, continue to step 9.
Several to many different multifusion plasmid combinations can be processed and
selected on one 96 well microtiter plate in parallel.
9. For 96 well antibiotic tests, inoculate four colonies from each agar plate with
different antibiotic combination into 500 µl LB media without antibiotics.
Incubate the cell cultures in a 37°C shaking incubator for 1-2 hours.
ACEMBL System User Manual
EMBL Grenoble, 2009
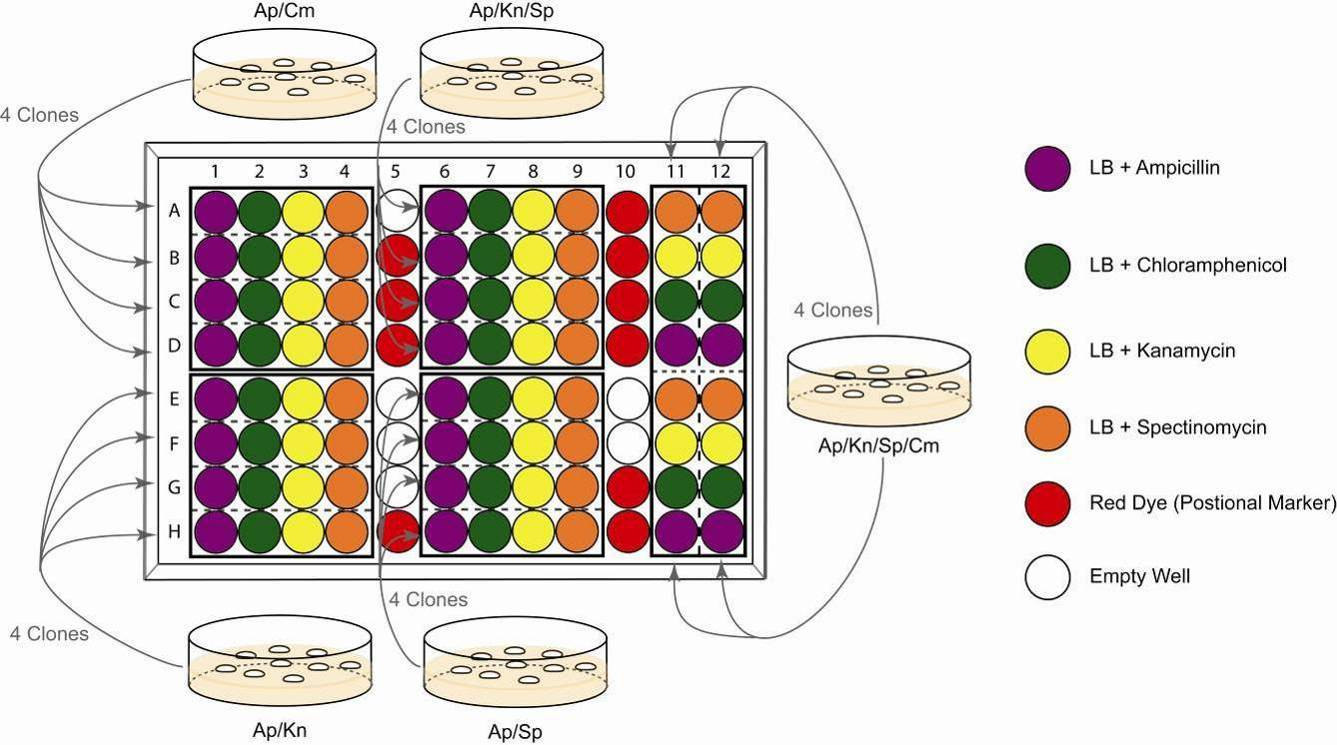
10. During the incubation of colonies, fill a 96 well microtiter plate with 150 µl
antibiotic-containing LB media (following Illustration 7). It is recommended
to add coloured dye (positional marker) in the wells indicated.
A typical arrangement of the solutions, which is used for parallel selections of
multifusion plasmids, is shown in Illustration 7. The concept behind the 96 well plate
experiment is that every cell suspension from single colonies needs to be challenged
by all four single antibiotics for unambiguous interpretation.
Illustration 7: 96 well analysis of Cre assembly
11. Add 1 µl aliquots of pre-incubated cell culture (Step 9) to the corresponding
wells. Then incubate the inoculated 96 well microtiter plate in a 37°C shaking
incubator overnight at 180-200 rpm.
Recommended: use parafilm to wrap the plate to avoid drying out.
The remainder of the pre-incubated cell cultures could be kept at 4°C for further
inoculations if necessary.
12. Select transformants containing desired multifusion plasmids based on
antibiotic resistance, according to the combination of dense (positive) and
clear (no growth) cell microcultures from each colony. Inoculate 10-20 µl cell
culture into 10 ml LB media with corresponding antibiotics. Incubate in a
37°C shaking incubator overnight.
13. Centrifuge the overnight cell cultures at 4000g for 5-10 minutes. Purify
plasmid from the resulting cell pellets with common plasmid miniprep kits,
according to manufacturers' recommendation.
ACEMBL System User Manual
EMBL Grenoble, 2009
14. Determine the concentrations of purified plasmid solutions by using UV
absorption spectroscopy (e.g. by using a NanoDropTM 1000 machine).
15. Digest 0.5 1 µg of the purified plasmid solution in a 20 µl restriction
digestion with appropriate endonuclease(s). Incubate under recommended
reaction condition for 2 hours.
16. Use 5-10 µl of the digestion for analytical agarose (0.8-1.2%) gel
electrophoresis. Verify plasmid integrity by comparing the experimental
restriction pattern to a restriction pattern predicted in silico (e.g. by using
program VectorNTI from Invitrogen or similar programs).
C.2.2. Deconstruction of fusion vectors by Cre
The following protocol can be used for example also for the recovery of all four single ACEMBL vectors by deconstructing tetra-fused pACKS plasmid (pACE-pDC-pDK-pDS); which is part of the ACEMBL System kit (Section D). Likewise, the protocol is suitable for releasing any single educt from multifusion constructs (deconstruction). This is achieved by Cre-LoxP reaction, transformation and plating on agar with appropriately reduced antibiotic resistance level (c.f. Illustration 6). In the liberated educt entity, encoding genes can be modified and diversified. Then, the diversified construct is resupplied by Cre-LoxP reaction (C.2.1.).
Cre recombinase (and 10x Buffer) E. coli competent cells
(pir+ strains, pir- strains could be used only when partially deconstructed Acceptor-Donor fusions are desired).
1. Incubate 1 µg multifusion plasmid with 2 µl 10x Cre buffer, 1 2 µl Cre
recombinase, add ddH2O to adjust the total reaction volume to 20 µl.
2. Incubate this Cre deconstruction reaction mixture at 30°C for 1-4 hour. 3. Optional: load 2-5 µl of the reaction on an analytical agarose gel for
examination. Heat inactivation at 70°C for 10 minutes before the gel loading is strongly
recommended.
4. For chemical transformation, mix 10-15µl De-Cre reaction with 200 µl
chemical competent cells. Incubate the mixture on ice for 15-30 minutes.
Then perform heat shock at 42°C for 45-60 s. Up to 20 µl De-Cre reaction (0.1 volumes of the chemical competent cell suspension)
can be directly transformed into 200 µl chemical competent cells.
ACEMBL System User Manual
EMBL Grenoble, 2009
For electrotransformation, up to 2 µl De-Cre reaction could be directly mixed
with 100 µl electrocompetent cells, and transformed by using an
electroporator (e.g. BIORAD E. coli Pulser) at 1.8-2.0 kV. Larger volume of De-Cre reaction must be desalted by ethanol precipitation or PCR
purification column before electrotransformation. The desalted De-Cre reaction mix
should not exceed 0.1 volumes of the electrocompetent cell suspension. The cell/DNA mixture could be immediately used for electrotransformation without
prior incubation on ice.
5. Add up to 400 µl of LB media (or SOC media) per 100 µl of cell/DNA
suspension immediately after the transformation (heat shock or
electroporation).
6. Incubate the suspension in a 37°C shaking incubator (recovery).
For recovery of partially deconstructed double/triple fusions, incubate the
suspension in a 37°C shaking incubator for 1 to 2 hours. For recovery of individual educts such as single ACEMBL vectors from pACKS
plasmid, incubate the suspension in a 37°C whaking incubator overnight or for at
least 4 hours.
7. Plate out the recovered cell suspension on agar containing the desired
(combination of) antibiotic(s). Incubate at 37°C overnight.
8. Colonies after overnight incubation might be verified directly by restriction
digestion at this stage (refer to steps 12-16). Especially recommended in the case that only one single educt or partially
deconstructed multifusion plasmid is desired. For further selection by single antibiotic challenge on a 96 well microtiter
plate, continue with step 9. Several different single educts/partially deconstructed multifusion plasmids can be
processed and selected on one 96 well microtiter plate in parallel.
9. For 96 well analysis, inoculate four colonies each from agar plates containing
a defined set of antibiotics into 500 µl LB media without antibiotics.
Incubate the cell cultures in a 37°C shaking incubator for 1-2 hours.
10. During the incubation of colonies, fill a 96 well microtiter plate with 150 µl
antibiotic-containing LB media or coloured dye (positional marker) in the
corresponding wells. Refer to Illustrations 7 and 12 for the arrangement of the solutions in the wells,
which are used for parallel selection of single educts or partially deconstructed
multifusion plasmids. The concept is that every cell suspension from a single colony
needs to be challenged by all four antibiotics separately for unambiguous
interpretation.
11. Add 1 µl aliquots from the pre-incubated cell cultures (Step 9) into the
corresponding wells. Then incubate the 96 well microtiter plate in a 37°C
shaking incubator overnight at 180-200 rpm.
Recommended: use parafilm to wrap the plate to prevent dehydration.
The remainder of the pre-incubated cell cultures can be kept in 4°C fridge for further
inoculations if necessary.
ACEMBL System User Manual
EMBL Grenoble, 2009
12. Select transformants containing desired single educts or partially
deconstructed multifusion plasmids according to the combination of dense
(growth) and clear (no growth) cell cultures from each colony. Inoculate 10-
20 µl cell cultures into 10 ml LB media with corresponding antibiotic(s).
Incubate in a 37°C shaking incubator overnight.
13. Centrifuge the overnight cell cultures at 4000g for 5-10 minutes. Purify
plasmid from cell pellets with common plasmid miniprep kits, according to
manufacturers' information.
14. Determine the concentrations of purified plasmid solutions by using UV
absorption spectroscopy (e.g. NanoDropTM 1000).
15. Digest 0.5 1 µg of the purified plasmid solution in a 20 µl restriction
digestion (with 5-10 unit endonuclease). Incubate under recommended
reaction condition for 2 hours.
16. Use 5-10 µl of the digestion for analytical agarose gel (0.8-1.2%)
electrophoresis. Verify the plasmid integrity by comparing the actual
restriction pattern to predicted restriction pattern in silico (e.g. by using
VectorNTI, Invitrogen, or any other similar program).
17. Optional: Possibly, a deconstruction reaction is not complete but yields
partially deconstructed fusions which still retain entities to be eliminated. In
this case, we recommend to pick these partially deconstructed fusions
containing and perform a second round of Cre deconstruction reaction (repeat
steps 1-8) by using this construct as starting material. In our hands, two sequential deconstruction reactions were always sufficient to
recover all individual modules, for instance all four single ACEMBL vectors from a
pACKS plasmid. Liberation of single educts from double/triple fusions were found to
be often more efficient than from quadruples such as the pACKS plasmid of the
system kit (Section E).
C.3. Coexpression by Cotransformation
Protein complexes can be expressed also from two separate vectors that were cotransformed in expression strains. The cotransformed vectors can have the same or different origins of replication, however, they must encode for different resistance markers. Plasmids pACE (ampicillin resistance marker) and pACE2 (tetracycline resistance marker) have both a ColE1 derived replicon and can therefore be used with all common expression strains. pACE and pACE2 derivatives (also including fused Donors if needed) can be cotransformed into expression strains, and double transformants selected for by plating on agar plates containing both ampicillin and tetracyclin antibiotics.
Transformations are carried out by using standard transformation protocols.
ACEMBL System User Manual
EMBL Grenoble, 2009
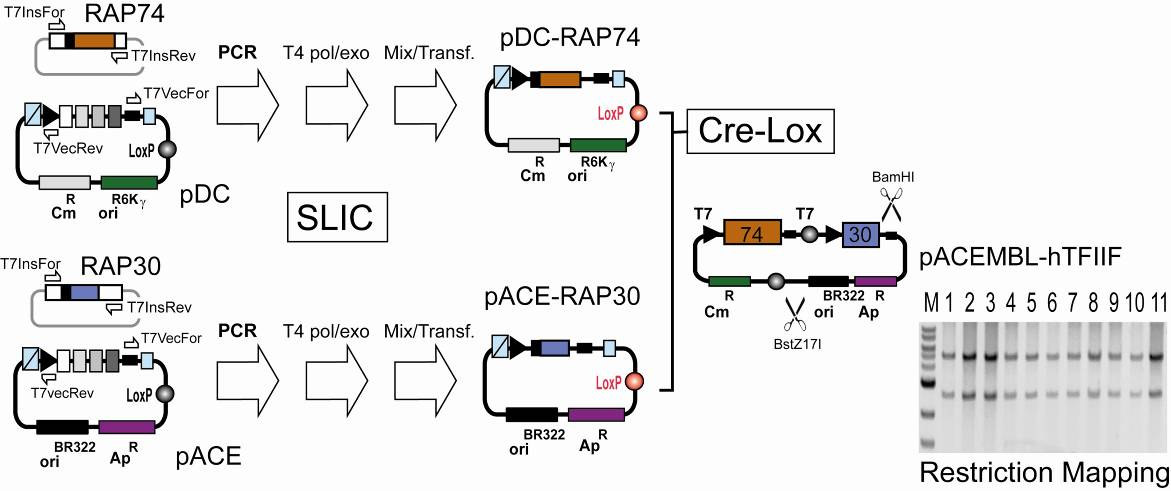
D. ACEMBL multigene combination: Examples Examples of multiprotein expressions by ACEMBL are shown in the following illustrating the gene combination procedures detailed in Section C. Reactions presented were carried out manually following the protocols provided, and also on a Tecan Freedom EvoII 200 robot with adapted protocols. D.1. SLIC cloning into ACEMBL vectors: human TFIIF Genes encoding for full-length human RAP74 with a C-terminal oligo-histidine tag and full-length human RAP30 were amplified from pET-based plasmid template9 by using the primer pair TN7InsFor (5'-TCCCGCGAAATTAATACGACTCACTATA GGG-3') and Tn7Insrev (5'-CCTCAAGACCCGTTTAGAGGCCCCAAGGGGTT ATGCTAG-3') following the protocols described above. Linearized vector backbones were generated by PCR amplification from pACE and pDC by using primer pair Tn7VecFor (5'CTAGCATAACCCCTTGGGGCCTCTAAACGGGT CTTGAGG-3') and Tn7VecRev (5'-CCCTATAGTGAGTCGTATTAATTTC GCGGGA-3') in both cases. SLIC following Protocol 1 (Section C), resulting in pACE-RAP30 and pDC-RAP74his (Fig 8). These plasmids were fused by Cre-LoxP reaction (Section C). Results from restriction mapping by BstZ17I/BamHI double digestion of 11 double resistant (Cm, Ap) colonies are shown by a gel section from 1% E-gel electrophoresis (M: NEB 1kb DNA marker). All clones tested showed the expected pattern (5.0 + 2.8 kb). One clone was transformed in BL21(DE3) cells. Expression and purification by Ni2+-capture and S200 chromatography resulted in human TFIIF complex (Fig. 3a, main text).
Illustration 8: ACEMBLing TFIIF.
9 Gaiser, F., Tan, S. and Richmond, T.J. J. Mol. Biol. 302, 1119-1127 (2000).
ACEMBL System User Manual
EMBL Grenoble, 2009
D.2. Polycistron by SLIC: human VHL/ElonginB/ElonginC complex. The gene encoding for Von Hippel Lindau protein (amino acids 54-213), fused at its N-terminus to a six-histidine-thioredoxin fusion tag, was PCR amplified from plasmid pET3-HisTrxVHL by using primers Tn7InsFor (Table I) and SmaBamVHL (5'-GAATTCACTGGCCGTCGTTTTACAGGATCCTTAATCTCCCATCCGTTG ATGTGCAATG-3'). SmaBamVHL primer is a derivative of the SmaBam adaptor sequence (Table I) elongated at its 3' by the insert specific sequence at the 3' end of the VHL gene (including a stop codon). The gene encoding for full-length ElonginB was PCR amplified from pET3-ElonginB by using primers BamSmaEB (5'-GGATCCTGTAAAACGACGGCCAGTGAATTCGCTAGCTCTAGAAATAATTTGTTTAAC-3') and SacHindEB (5'-GAGCTCGACTGGGAAAACCCTGGCG AAGCTTAGATCTGGATCCTTACTGCACGGCTTGTTCATTGG-3'), which are derivatives of the corresponding adaptors (Table I). The gene for ElonginC (amino acids 17-112) wa amplified from pET3-ElonginC by using primers HindSacEC (5'-AAGCTTCGCCAGGGTTTTCCCAGTCGAGCTCCAATTGGAATTCGCTAGCTCTAG-3') and BspEco5EC (5'GATCCGGATGTGAAATTGTTATCCGCTGG TACCAAGCTTAGATCTGGATCCTTAACAATCTAAGAAG-3'), which are derivatives of the corresponding adaptors (Table I). Vector backbone was PCR amplified by using primers Tn7VecRev and Eco5Bsp, and pACE as a template (Illustr. 9). Multifragment SLIC was carried out according to Protocol 2 (Section C) resulting in pACE-VCB which contains a tricistron. Clones were plated on agar plates containing ampicillin. A positive clone, verified by sequencing, was used in the coexpression experiment described below (section D.5.)
Illustration 9: Multifragment SLIC of pACE-VHLbc (tricistron).
ACEMBL System User Manual
EMBL Grenoble, 2009
D.3. The Homing endonuclease/BstXI module: yeast RES complex Plasmids pCDFDuet-Pml1p, pRSFDuet-Snu17p-NHis and pETDuet-Bud13p, encoding for yeast proteins (all full-length) PmI1p, Snu17p and Bud13p, respectively, were a kind gift from Dr. Simon Trowitzsch and Dr. Markus Wahl (MPI Göttingen). Snu17p contains a six-histidine tag fused to its N-terminus. The gene encoding for His6-tagged Snu17p was excised from pRSFDuet-Snu17p-NHis by using restriction enzymes NcoI and XhoI, and ligated into a NcoI/XhoI digested pACE construct (containing an unrelated gene between NcoI and XhoI sites) resulting in pACE-Snu17. The gene encoding for Bud13p was liberated from pETDuet-Bud13p by restriction digestion with XbaI and EcoRV, and placed into XbaI/PmeI digested pDC resulting in pDC-Bud13. The gene encoding for Pm1Ip was liberated from pCDFDuet-Pml1p by restriction digestion with NdeI and XhoI, and placed into NdeI/XhoI digested pDC resulting in pDC-PmI1. Next, the expression cassette for Bud13p was liberated from pDC-Bud13 by digestion with PI-SceI and BstXI. The liberated fragment was inserted into PI-SceI digested and alkaline phosphatase treated pDC-PmI1p resulting in pDC-Bud13p-PmI1p.
pACE-Snu17 and pDC-BudPmI were then fused by Cre-LoxP reaction and
selected for by plating on agar plates containing ampicillin and chloramphenicol. Fusion plasmids were transformed into BL21(DE3) cells. Expression and purification by Ni2+-capture and S200 size exclusion chromatography resulted in the trimeric RES complex (Supplementary Results, complex S12b).
Illustration 10: The HE/BstXI multiplication module.
ACEMBL System User Manual
EMBL Grenoble, 2009
D.4. Coexpression by cotransformation: human NYB/NYC Genes encoding for protein NYB (amino acids 49-141) and NYC (amino acids 27-12) were excised from vectors pACYC18411-NYB and pET15-NYC, respectively10. NdeI and BamHI where used for NFYB. XbaI and BamHI where used for NYC, thus importing a six-histidine tag at the N-terminus of the protein. The NYB insert was ligated into pACE digested with NdeI and BamHI. The NYC insert was ligated into pACE2 digested by XbaI and BamHI. pACE-NFYB and pACE2-NFYC were transformed into BL21(DE3) cells containing the pLysS plasmid. Selection on agar plates containing ampicillin, tetracyclin and chloramphenicol resulted in triple resistant colonies. The complex was expressed and purified by Ni2+ capture (IMAC) and S75HR (Pharmacia) size exclusion chromatography (Supplementary Results, complex S7a). D.5. Coexpression from Acceptor-Donor fusions Six heterologous genes encoding for a trimeric protein complex (VHLbc: VonHippel-Lindau protein amino acids 54-213 / full-length ElonginB / ElonginC amino acids 17-112)11, a gene encoding for the AAA ATPase FtsH (amino acids 147-610), and two genes encoding for fluorescent markers (BFP and GFP) were assembled as indicated. In a single Cre reaction, all combinations of one Acceptor (pACE-VHLbc) and three Donors (pDC-FtsH, pDK-BFP, pDS-mGFP) were obtained and selected, including a quadruple fusion containing all six heterologous genes (Main text, Fig. 2). Clones were verified by 96 well microtiter assay as described in Section C. Expression and Ni2+ affinity capture, combined with immunostaining of the untagged fluorescent markers, confirmed successful multiprotein expression (Main text, Figs. 2 and 3b). Proteins were expressed overnight in BL21(DE3) cells in 24 well deep-well plates in small scale using autoinduction media12. Restriction mapping revealed that even large fusion plasmids were stable over many (more than 60) generations, even if challenged by a single antibiotic in the medium only.
10 Romier, C. et al., J. Biol. Chem. 278, 1336-1345 (2003)
11 Stebbins, C.E., Kaelin, W.G. Jr, Pavletich, N.P. Science 284, 455-61 (1999)
12 Studier F.W. Protein Expr. Purif. 41, 207-34 (2005).
ACEMBL System User Manual
EMBL Grenoble, 2009
E. The ACEMBL System Kit
Reagents to be supplied in ACEMBL system kit:
BW23473, BW23474 cells†
pACKS quadruple fusion vector* made of: pACE (Acceptor)
pDC, pDK, pDS (Donors)
pACE-[VHLbc/BFP/mGFP] control plasmid triple fusion vector made of: pACE-VHLbc
† E. coli strains expressing the pir gene for propagation of Donor
derivatives (any other strain with pir+ background can be used).
* This fusion vector was created by Cre-LoxP reaction of pACE, pDC,
pDK and pDS. It is resistant to ampicillin, kanamycin, chloramphenicol and spectinomycin. Individual ACEMBL vectors are liberated from this quadruple fusion by Cre-LoxP mediated deconstruction as described in protocol C.2.2. Sequences for single ACEMBL vectors and pACKS quadruple fusion are provided in Appendix.
# pDS-mGFP contains a coiled-coil fused to the N-terminus of eGFP13.
Reagents additionally required:
Antibiotics: ampicillin, chloramphenicol, kanamycin, spectinomycin, tetracycline
Enzymes: Cre recombinase
T4 DNA polymerase (for recombination insertion of genes) Phusion polymerase (for PCR amplification of DNA) Restriction enzymes and T4 DNA ligase (for conventional cloning)
Regular laboratory cloning strain (TOP10, HB101, DH5α)
Expression strain(s) of choice
13 Berger, P. et al., Proc. Natl. Acad. Sci. USA 100, 12177-82 (2003).
ACEMBL System User Manual
EMBL Grenoble, 2009
Illustration 11: ACEMBL System Kit: Generating single vectors from pACKS.
pACKS is deconstructed according to the schematic in Illustr. 11 into single vectors pACE, pDC, pDK and pDS. 96 well microtiter assay for identifying single vectors is shown in Illustr. 12.
ACEMBL System User Manual
EMBL Grenoble, 2009
Illustration 12: 96 well microtiter analysis of pACKS De-Cre reaction.
Clones containing pACE, pDC, pDK and pDS single vectors as identified by
microtiter assay, are then used for plasmid generation. The vectors can be further verified by restriction digestion before use for subcloning (see Appendix for vector sequences). pACE2 is provided as a separate vector in the ACEMBL System Kit.
ACEMBL System User Manual
EMBL Grenoble, 2009
F. Appendix
F.1. DNA sequence of MIE
Below are the sequence and map of the MIE fragment between T7/lac promoter and
T7 terminator in ACEMBL vectors. Forward and reverse primers for sequencing can
be standard vector primers for T7 and lac. Adaptor primer sequences (c.f. Table I) are
indicated. DNA sequences in these homology regions contain tried-and-tested
sequencing primers14. Sites of insertion (I1-I4) are shown. The adaptor sequences,
and probably any sequence in the homology regions, can be used as adaptors for
multifragment insertions. The ribosome binding site present in the MIE (rbs) is boxed
in red.
14 Tan S. et al. Protein Expr. Purif. 40, 385, (2005).
ACEMBL System User Manual
EMBL Grenoble, 2009
F.2. DNA sequences of ACEMBL vectors F.2.1. pACE 1
GGTACCGCGG CCGCGTAGAG GATCTGTTGA TCAGCAGTTC AACCTGTTGA
TAGTACTTCG TTAATACAGA TGTAGGTGTT GGCACCATGC ATAACTATAA
CGGTCCTAAG GTAGCGACCT AGGTATCGAT AATACGACTC ACTATAGGGG
AATTGTGAGC GGATAACAAT TCCCCTCTAG AAATAATTTT GTTTAACTTT
AAGAAGGAGA TATACATATG AGGCCTCGGA TCCTGTAAAA CGACGGCCAG
TGAATTCCCC GGGAAGCTTC GCCAGGGTTT TCCCAGTCGA GCTCGATATC
GGTACCAGCG GATAACAATT TCACATCCGG ATCGCGAACG CGTCTCGAGA
GATCCGGCTG CTAACAAAGC CCGAAAGGAA GCTGAGTTGG CTGCTGCCAC
CGCTGAGCAA TAACTAGCAT AACCCCTTGG GGCCTCTAAA CGGGTCTTGA
GGGGTTTTTT GGTTTAAACC CATCTAATTG GACTAGTAGC CCGCCTAATG
AGCGGGCTTT TTTTTAATTC CCCTATTTGT TTATTTTTCT AAATACATTC
AAATATGTAT CCGCTCATGA GACAATAACC CTGATAAATG CTTCAATAAT
ATTGAAAAAG GAAGAGTATG AGTATTCAAC ATTTCCGTGT CGCCCTTATT
CCCTTTTTTG CGGCATTTTG CCTTCCTGTT TTTGCTCACC CAGAAACGCT
CGTGAAAGTA AAAGACGCAG AGGACCAATT GGGGGCACGA GTGGGATACA
TAGAACTGGA CTTGAATAGC GGTAAAATCC TTGAGAGTTT TCGCCCTGAA
GAGCGTTTTC CAATGATGAG CACTTTCAAA GTTCTGCTAT GTGGAGCAGT
ATTATCCCGT GTAGATGCGG GGCAAGAGCA ACTCGGACGA CGAATACACT
ATTCGCAGAA TGACTTGGTT GAATACTCCC CAGTGACAGA AAAGCACCTT
ACGGACGGAA TGACGGTAAG AGAATTATGT AGTGCCGCCA TAACGATGAG
TGATAACACT GCGGCGAACT TACTTCTGAC AACCATCGGT GGACCGAAGG
AATTAACCGC TTTTTTGCAC AATATGGGAG ACCATGTAAC TCGCCTTGAC
CGTTGGGAAC CAGAACTGAA TGAAGCCATA CCAAACGACG AGCGAGACAC
CACAATGCCT GCGGCAATGG CAACAACATT ACGCAAACTA TTAACTGGCG
AACTACTTAC TCTGGCTTCA CGGCAACAAT TAATAGACTG GCTTGAAGCG
GATAAAGTTG CAGGACCACT ACTGCGTTCG GCACTTCCTG CTGGCTGGTT
TATTGCTGAT AAATCTGGGG CAGGAGAGCG TGGTTCACGG GGTATCATTG
CCGCACTTGG ACCAGATGGT AAGCCTTCCC GTATCGTAGT TATCTACACG
ACGGGTAGTC AGGCAACTAT GGACGAACGA AATAGACAGA TTGCTGAAAT
AGGGGCTTCA CTGATTAAGC ATTGGTAAAC CGATACAATT AAAGGCTCCT
TTTGGAGCCT TTTTTTTTGG ACGGACCGGT AGAAAAGATC AAAGGATCTT
CTTGAGATCC TTTTTTTCTG CGCGTAATCT GCTGCTTGCA AACAAAAAAA
CCACCGCTAC CAGCGGTGGT TTGTTTGCCG GATCAAGAGC TACCAACTCT
TTTTCCGAAG GTAACTGGCT TCAGCAGAGC GCAGATACCA AATACTGTCC
TTCTAGTGTA GCCGTAGTTA GGCCACCACT TCAAGAACTC TGTAGCACCG
CCTACATACC TCGCTCTGCT AATCCTGTTA CCAGTGGCTG CTGCCAGTGG
CGATAAGTCG TGTCTTACCG GGTTGGACTC AAGACGATAG TTACCGGATA
AGGCGCAGCG GTCGGGCTGA ACGGGGGGTT CGTGCACACA GCCCAGCTTG
GAGCGAACGA CCTACACCGA ACTGAGATAC CTACAGCGTG AGCTATGAGA
AAGCGCCACG CTTCCCGAAG GGAGAAAGGC GGACAGGTAT CCGGTAAGCG
GCAGGGTCGG AACAGGAGAG CGCACGAGGG AGCTTCCAGG GGGAAACGCC
TGGTATCTTT ATAGTCCTGT CGGGTTTCGC CACCTCTGAC TTGAGCGTCG
ATTTTTGTGA TGCTCGTCAG GGGGGCGGAG CCTATGGAAA AACGCCAGCA
ACGCGGCCTT TTTACGGTTC CTGGCCTTTT GCTGGCCTTT TGCTCACATG
TTCTTTCCTG CGTTATCCCC TGATTCTGTG GATAACCGTA TTACCGCCTT
TGAGTGAGCT GATACCGCTC GCCGCAGCCG AACGACCGAG CGCAGCGAGT
CAGTGAGCGA GGAAGCGGAA GAGCGCCTGA TGCGGTATTT TCTCCTTACG
CATCTGTGCG GTATTTCACA CCGCAATGGT GCACTCTCAG TACAATCTGC
TCTGATGCCG CATAGTTAAG CCAGTATACA CTCCGCTATC GCTACGTGAC
TGGGTCATGG CTGCGCCCCG ACACCCGCCA ACACCCGCTG ACGCGCCCTG
ACGGGCTTGT CTGCTCCCGG CATCCGCTTA CAGACAAGCT GTGACCGTCT
CCGGGAGCTG CATGTGTCAG AGGTTTTCAC CGTCATCACC GAAACGCGCG
AGGCAGGGGG AATTCCAGAT AACTTCGTAT AATGTATGCT ATACGAAGTT
ACEMBL System User Manual
EMBL Grenoble, 2009
ATGAAATCTA ACAATGCGCT CATCGTCATC CTCGGCACCG TCACCCTGGA
TGCTGTAGGC ATAGGCTTGG TTATGCCGGT ACTGCCGGGC CTCTTGCGGG
ATATCGTCCA TTCCGACAGC ATCGCCAGTC ACTATGGCGT GCTGCTAGCG
CTATATGCGT TGATGCAATT TCTATGCGCA CCCGTTCTCG GAGCACTGTC
CGACCGCTTT GGCCGCCGCC CAGTCCTGCT CGCTTCGCTA CTTGGAGCCA
CTATCGACTA CGCGATCATG GCGACCACAC CCGTCCTGTG GATTCTCTAC
GCCGGACGCA TCGTGGCCGG CATCACCGGC GCCACAGGTG CGGTTGCTGG
CGCCTATATC GCCGACATCA CCGATGGGGA AGATCGGGCT CGCCACTTCG
GGCTCATGAG CGCTTGTTTC GGCGTGGGTA TGGTGGCAGG CCCCGTGGCC
GGGGGACTGT TGGGCGCCAT CTCCTTACAT GCACCATTCC TTGCGGCGGC
GGTGCTCAAC GGCCTCAACC TACTACTGGG CTGCTTCCTA ATGCAGGAGT
CGCATAAGGG AGAGCGCCGA CCCATGCCCT TGAGAGCCTT CAACCCAGTC
AGCTCCTTCC GGTGGGCGCG GGGCATGACT ATCGTCGCCG CACTTATGAC
TGTCTTCTTT ATCATGCAAC TCGTAGGACA GGTGCCGGCA GCGCTCTGGG
TCATTTTCGG CGAGGACCGC TTTCGCTGGA GCGCGACGAT GATCGGCCTG
TCGCTTGCGG TATTCGGAAT CTTGCACGCC CTCGCTCAAG CCTTCGTCAC
TGGTCCCGCC ACCAAACGTT TCGGCGAGAA GCAGGCCATT ATCGCCGGCA
TGGCGGCCGA CGCGCTGGGC TACGTCTTGC TGGCGTTCGC GACGCGAGGC
TGGATGGCCT TCCCCATTAT GATTCTTCTC GCTTCCGGCG GCATCGGGAT
GCCCGCGTTG CAGGCCATGC TGTCCAGGCA GGTAGATGAC GACCATCAGG
GACAGCTTCA AGGATCGCTC GCGGCTCTTA CCAGCCTAAC TTCGATCATT
GGACCGCTGA TCGTCACGGC GATTTATGCC GCCTCGGCGA GCACATGGAA
CGGGTTGGCA TGGATTGTAG GCGCCGCCCT ATACCTTGTC TGCCTCCCCG
CGTTGCGTCG CGGTGCATGG AGCCGGGCCA CCTCGACCTG AACCGATACA
ATTAAAGGCT CCTTTTGGAG CCTTTTTTTT TGGACGGACC GGTAGAAAAG
ATCAAAGGAT CTTCTTGAGA TCCTTTTTTT CTGCGCGTAA TCTGCTGCTT
GCAAACAAAA AAACCACCGC TACCAGCGGT GGTTTGTTTG CCGGATCAAG
AGCTACCAAC TCTTTTTCCG AAGGTAACTG GCTTCAGCAG AGCGCAGATA
CCAAATACTG TCCTTCTAGT GTAGCCGTAG TTAGGCCACC ACTTCAAGAA
CTCTGTAGCA CCGCCTACAT ACCTCGCTCT GCTAATCCTG TTACCAGTGG
CTGCTGCCAG TGGCGATAAG TCGTGTCTTA CCGGGTTGGA CTCAAGACGA
TAGTTACCGG ATAAGGCGCA GCGGTCGGGC TGAACGGGGG GTTCGTGCAC
ACAGCCCAGC TTGGAGCGAA CGACCTACAC CGAACTGAGA TACCTACAGC
GTGAGCTATG AGAAAGCGCC ACGCTTCCCG AAGGGAGAAA GGCGGACAGG
TATCCGGTAA GCGGCAGGGT CGGAACAGGA GAGCGCACGA GGGAGCTTCC
AGGGGGAAAC GCCTGGTATC TTTATAGTCC TGTCGGGTTT CGCCACCTCT
GACTTGAGCG TCGATTTTTG TGATGCTCGT CAGGGGGGCG GAGCCTATGG
AAAAACGCCA GCAACGCGGC CTTTTTACGG TTCCTGGCCT TTTGCTGGCC
TTTTGCTCAC ATGTTCTTTC CTGCGTTATC CCCTGATTCT GTGGATAACC
GTATTACCGC CTTTGAGTGA GCTGATACCG CTCGCCGCAG CCGAACGACC
GAGCGCAGCG AGTCAGTGAG CGAGGAAGCG GAAGAGCGCC TGATGCGGTA
TTTTCTCCTT ACGCATCTGT GCGGTATTTC ACACCGCAAT GGTGCACTCT
CAGTACAATC TGCTCTGATG CCGCATAGTT AAGCCAGTAT ACACTCCGCT
ATCGCTACGT GACTGGGTCA TGGCTGCGCC CCGACACCCG CCAACACCCG
CTGACGCGCC CTGACGGGCT TGTCTGCTCC CGGCATCCGC TTACAGACAA
GCTGTGACCG TCTCCGGGAG CTGCATGTGT CAGAGGTTTT CACCGTCATC
ACCGAAACGC GCGAGGCAGG GGGAATTCCA GATAACTTCG TATAATGTAT
GCTATACGAA GTTATGGTAC CGCGGCCGCG TAGAGGATCT GTTGATCAGC
AGTTCAACCT GTTGATAGTA CTTCGTTAAT ACAGATGTAG GTGTTGGCAC
CATGCATAAC TATAACGGTC CTAAGGTAGC GACCTAGGTA TCGATAATAC
GACTCACTAT AGGGGAATTG TGAGCGGATA ACAATTCCCC TCTAGAAATA
ATTTTGTTTA ACTTTAAGAA GGAGATATAC ATATGAGGCC TCGGATCCTG
TAAAACGACG GCCAGTGAAT TCCCCGGGAA GCTTCGCCAG GGTTTTCCCA
GTCGAGCTCG ATATCGGTAC CAGCGGATAA CAATTTCACA TCCGGATCGC
GAACGCGTCT CGAGAGATCC GGCTGCTAAC AAAGCCCGAA AGGAAGCTGA
GTTGGCTGCT GCCACCGCTG AGCAATAACT AGCATAACCC CTTGGGGCCT
CTAAACGGGT CTTGAGGGGT TTTTTGGTTT AAACCCATCT AATTGGACTA
GTAGCCCGCC TAATGAGCGG GCTTTTTTTT AATTCCCCTA TTTGTTTATT
TTTCTAAATA CATTCAAATA TGTATCCGCT CATGAGACAA TAACCCTGAT
AAATGCTTCA ATAATATTGA AAAAGGAAGA GT
ACEMBL System User Manual
EMBL Grenoble, 2009
ATCAACGTCT CATTTTCGCC AAAAGTTGGC CCAGATCTAT GTCGGGTGCG
GAGAAAGAGG TAATGAAATG GCACCTAGGT ATCGATAATA CGACTCACTA
TAGGGGAATT GTGAGCGGAT AACAATTCCC CTCTAGAAAT AATTTTGTTT
AACTTTAAGA AGGAGATATA CATATGAGGC CTCGGATCCT GTAAAACGAC
GGCCAGTGAA TTCCCCGGGA AGCTTCGCCA GGGTTTTCCC AGTCGAGCTC
GATATCGGTA CCAGCGGATA ACAATTTCAC ATCCGGATCG CGAACGCGTC
TCGAGAGATC CGGCTGCTAA CAAAGCCCGA AAGGAAGCTG AGTTGGCTGC
TGCCACCGCT GAGCAATAAC TAGCATAACC CCTTGGGGCC TCTAAACGGG
TCTTGAGGGG TTTTTTGGTT TAAACCCATG TGCCTGGCAG ATAACTTCGT
ATAATGTATG CTATACGAAG TTATGGTACC GCGGCCGCGT AGAGGATCTG
TTGATCAGCA GTTCAACCTG TTGATAGTAC GTACTAAGCT CTCATGTTTC
ACGTACTAAG CTCTCATGTT TAACGTACTA AGCTCTCATG TTTAACGAAC
TAAACCCTCA TGGCTAACGT ACTAAGCTCT CATGGCTAAC GTACTAAGCT
CTCATGTTTC ACGTACTAAG CTCTCATGTT TGAACAATAA AATTAATATA
AATCAGCAAC TTAAATAGCC TCTAAGGTTT TAAGTTTTAT AAGAAAAAAA
AGAATATATA AGGCTTTTAA AGCTTTTAAG GTTTAACGGT TGTGGACAAC
AAGCCAGGGA TGTAACGCAC TGAGAAGCCC TTAGAGCCTC TCAAAGCAAT
TTTGAGTGAC ACAGGAACAC TTAACGGCTG ACAGAATTAG CTTCACGCTG
CCGCAAGCAC TCAGGGCGCA AGGGCTGCTA AAGGAAGCGG AACACGTAGA
AAGCCAGTCC GCAGAAACGG TGCTGACCCC GGATGAATGT CAGCTGGGAG
GCAGAATAAA TGATCATATC GTCAATTATT ACCTCCACGG GGAGAGCCTG
AGCAAACTGG CCTCAGGCAT TTGAGAAGCA CACGGTCACA CTGCTTCCGG
TAGTCAATAA ACCGGTAAAC CAGCAATAGA CATAAGCGGC TATTTAACGA
CCCTGCCCTG AACCGACGAC CGGGTCGAAT TTGCTTTCGA ATTTCTGCCA
TTCATCCGCT TATTATCACT TATTCAGGCG TAGCAACCAG GCGTTTAAGG
GCACCAATAA CTGCCTTAAA AAAATTACGC CCCGCCCTGC CACTCATCGC
AGTACTGTTG TAATTCATTA AGCATTCTGC CGACATGGAA GCCATCACAA
ACGGCATGAT GAACCTGAAT CGCCAGCGGC ATCAGCACCT TGTCGCCTTG
CGTATAATAT TTGCCCATGG TGAAAACGGG GGCGAAGAAG TTGTCCATAT
TGGCCACGTT TAAATCAAAA CTGGTGAAAC TCACCCAGGG ATTGGCTGAG
ACGAAAAACA TATTCTCAAT AAACCCTTTA GGGAAATAGG CCAGGTTTTC
ACCGTAACAC GCCACATCTT GCGAATATAT GTGTAGAAAC TGCCGGAAAT
CGTCGTGGTA TTCACTCCAG AGCGATGAAA ACGTTTCAGT TTGCTCATGG
AAAACGGTGT AACAAGGGTG AACACTATCC CATATCACCA GCTCACCGTC
TTTCATTGCC ATACGGAATT CCGGATGAGC ATTCATCAGG CGGGCAAGAA
TGTGAATAAA GGCCGGATAA AACTTGTGCT TATTTTTCTT TACGGTCTTT
AAAAAGGCCG TAATATCCAG CTGAACGGTC TGGTTATAGG TACATTGAGC
AACTGACTGA AATGCCTCAA AATGTTCTTT ACGATGCCAT TGGGATATAT
CAACGGTGGT ATATCCAGTG ATTTTTTTCT CCATTTTAGC TTCCTTAGCT
CCTGAAAATC TCGATAACTC AAAAAATACG CCCGGTAGTG ATCTTATTTC
ATTATGGTGA AAGTTGGACC CTCTTACGTG CCGATCAACG TCTCATTTTC
GCCAAAAGTT GGCCCAG
ACEMBL System User Manual
EMBL Grenoble, 2009
CTATGTCGGG TGCGGAGAAA GAGGTAATGA AATGGCACCT AGGTATCGAT
GGCTTTACAC TTTATGCTTC CGGCTCGTAT GTTGTGTGGA ATTGTGAGCG
GATAACAATT TCACACAGGA AACAGCTATG ACCATGATTA CGAATTTCTA
GAAATAATTT TGTTTAACTT TAAGAAGGAG ATATACATAT GAGGCCTCGG
ATCCTGTAAA ACGACGGCCA GTGAATTCCC CGGGAAGCTT CGCCAGGGTT
TTCCCAGTCG AGCTCGATAT CGGTACCAGC GGATAACAAT TTCACATCCG
GATCGCGAAC GCGTCTCGAG ACTAGTTCCG TTTAAACCCA TGTGCCTGGC
AGATAACTTC GTATAATGTA TGCTATACGA AGTTATGGTA CGTACTAAGC
TCTCATGTTT CACGTACTAA GCTCTCATGT TTAACGTACT AAGCTCTCAT
GTTTAACGAA CTAAACCCTC ATGGCTAACG TACTAAGCTC TCATGGCTAA
CGTACTAAGC TCTCATGTTT CACGTACTAA GCTCTCATGT TTGAACAATA
AAATTAATAT AAATCAGCAA CTTAAATAGC CTCTAAGGTT TTAAGTTTTA
TAAGAAAAAA AAGAATATAT AAGGCTTTTA AAGCTTTTAA GGTTTAACGG
TTGTGGACAA CAAGCCAGGG ATGTAACGCA CTGAGAAGCC CTTAGAGCCT
CTCAAAGCAA TTTTCAGTGA CACAGGAACA CTTAACGGCT GACAGAATTA
GCTTCACGCT GCCGCAAGCA CTCAGGGCGC AAGGGCTGCT AAAGGAAGCG
GAACACGTAG AAAGCCAGTC CGCAGAAACG GTGCTGACCC CGGATGAATG
TCAGCTACTG GGCTATCTGG ACAAGGGAAA ACGCAAGCGC AAAGAGAAAG
CAGGTAGCTT GCAGTGGGCT TACATGGCGA TAGCTAGACT GGGCGGTTTT
ATGGACAGCA AGCGAACCGG AATTGCCAGC TGGGGCGCCC TCTGGTAAGG
TTGGGAAGCC CTGCAAAGTA AACTGGATGG CTTTCTTGCC GCCAAGGATC
TGATGGCGCA GGGGATCAAG ATCTGATCAA GAGACAGGAT GAGGATCGTT
TCGCATGATT GAACAAGATG GATTGCACGC AGGTTCTCCG GCCGCTTGGG
TGGAGAGGCT ATTCGGCTAT GACTGGGCAC AACAGACAAT CGGCTGCTCT
GATGCCGCCG TGTTCCGGCT GTCAGCGCAG GGGCGCCCGG TTCTTTTTGT
CAAGACCGAC CTGTCCGGTG CCCTGAATGA ACTGCAGGAC GAGGCAGCGC
GGCTATCGTG GCTGGCCACG ACGGGCGTTC CTTGCGCAGC TGTGCTCGAC
GTTGTCACTG AAGCGGGAAG GGACTGGCTG CTATTGGGCG AAGTGCCGGG
GCAGGATCTC CTGTCATCTC ACCTTGCTCC TGCCGAGAAA GTATCCATCA
TGGCTGATGC AATGCGGCGG CTGCATACGC TTGATCCGGC TACCTGCCCA
TTCGACCACC AAGCGAAACA TCGCATCGAG CGAGCACGTA CTCGGATGGA
AGCCGGTCTT GTCGATCAGG ATGATCTGGA CGAAGAGCAT CAGGGGCTCG
CGCCAGCCGA ACTGTTCGCC AGGCTCAAGG CGCGCATGCC CGACGGCGAG
GATCTCGTCG TGACACATGG CGATGCCTGC TTGCCGAATA TCATGGTGGA
AAATGGCCGC TTTTCTGGAT TCATCGACTG TGGCCGGCTG GGTGTGGCGG
ACCGCTATCA GGACATAGCG TTGGCTACCC GTGATATTGC TGAAGAGCTT
GGCGGCGAAT GGGCTGACCG CTTCCTCGTG CTTTACGGTA TCGCCGCTCC
CGATTCGCAG CGCATCGCCT TCTATCGCCT TCTTGACGAG TTCTTCTGAG
CGGGACTCTG GGGTTCGAAA TGACCGACCA AGCGACGCCC AACCTGCCAT
CACGAGATTT CGATTCCACC GCCGCCTTCT ATGAAAGGTT GGGCTTCGGA
ATCGTTTTCC GGGACGCCGG CTGGATGATC CTCCAGCGCG GGGATCTCAT
GCTGGAGTTC TTCGCCCACC CCGGGAT
ACEMBL System User Manual
EMBL Grenoble, 2009
CTATGTCGGG TGCGGAGAAA GAGGTAATGA AATGGCACCT AGGTATCGAT
GGCTTTACAC TTTATGCTTC CGGCTCGTAT GTTGTGTGGA ATTGTGAGCG
GATAACAATT TCACACAGGA AACAGCTATG ACCATGATTA CGAATTTCTA
GAAATAATTT TGTTTAACTT TAAGAAGGAG ATATACATAT GAGGCCTCGG
ATCCTGTAAA ACGACGGCCA GTGAATTCCC CGGGAAGCTT CGCCAGGGTT
TTCCCAGTCG AGCTCGATAT CGGTACCAGC GGATAACAAT TTCACATCCG
GATCGCGAAC GCGTCTCGAG ACTAGTTCCG TTTAAACCCA TGTGCCTGGC
AGATAACTTC GTATAATGTA TGCTATACGA AGTTATGGTA CGTACTAAGC
TCTCATGTTT CACGTACTAA GCTCTCATGT TTAACGTACT AAGCTCTCAT
GTTTAACGAA CTAAACCCTC ATGGCTAACG TACTAAGCTC TCATGGCTAA
CGTACTAAGC TCTCATGTTT CACGTACTAA GCTCTCATGT TTGAACAATA
AAATTAATAT AAATCAGCAA CTTAAATAGC CTCTAAGGTT TTAAGTTTTA
TAAGAAAAAA AAGAATATAT AAGGCTTTTA AAGCTTTTAA GGTTTAACGG
TTGTGGACAA CAAGCCAGGG ATGTAACGCA CTGAGAAGCC CTTAGAGCCT
CTCAAAGCAA TTTTGAGTGA CACAGGAACA CTTAACGGCT GACATAATTC
AGCTTCACGC TGCCGCAAGC ACTCAGGGCG CAAGGGCTGC TAAAGGAAGC
GGAACACGTA GAAAGCCAGT CCGCAGAAAC GGTGCTGACC CCGGATGAAT
GTCAGCTGGG AGGCAGAATA AATGATCATA TCGTCAATTA TTACCTCCAC
GGGGAGAGCC TGAGCAAACT GGCCTCAGGC ATTTGAGAAG CACACGGTCA
CACTGCTTCC GGTAGTCAAT AAACCGGTAA GTAGCGTATG CGCTCACGCA
ACTGGTCCAG AACCTTGACC GAACGCAGCG GTGGTAACGG CGCAGTGGCG
GTTTTCATGG CTTGTTATGA CTGTTTTTTT GGGGTACAGT CTATGCCTCG
GGCATCCAAG CAGCAAGCGC GTTACGCCGT GGGTCGATGT TTGATGTTAT
GGAGCAGCAA CGATGTTACG CAGCAGGGCA GTCGCCCTAA AACAAAGTTA
AACATCATGA GGGAAGCGGT GATCGCCGAA GTATCGACTC AACTATCAGA
GGTAGTTGGC GTCATCGAGC GCCATCTCGA ACCGACGTTG CTGGCCGTAC
ATTTGTACGG CTCCGCAGTG GATGGCGGCC TGAAGCCACA CAGTGATATT
GATTTGCTGG TTACGGTGAC CGTAAGGCTT GATGAAACAA CGCGGCGAGC
TTTGATCAAC GACCTTTTGG AAACTTCGGC TTCCCCTGGA GAGAGCGAGA
TTCTCCGCGC TGTAGAAGTC ACCATTGTTG TGCACGACGA CATCATTCCG
TGGCGTTATC CAGCTAAGCG CGAACTGCAA TTTGGAGAAT GGCAGCGCAA
TGACATTCTT GCAGGTATCT TCGAGCCAGC CACGATCGAC ATTGATCTGG
CTATCTTGCT GACAAAAGCA AGAGAACATA GCGTTGCCTT GGTAGGTCCA
GCGGCGGAGG AACTCTTTGA TCCGGTTCCT GAACAGGATC TATTTGAGGC
GCTAAATGAA ACCTTAACGC TATGGAACTC GCCGCCCGAC TGGGCTGGCG
ATGAGCGAAA TGTAGTGCTT ACGTTGTCCC GCATTTGGTA CAGCGCAGTA
ACCGGCAAAA TCGCGCCGAA GGATGTCGCT GCCGACTGGG CAATGGAGCG
CCTGCCGGCC CAGTATCAGC CCGTCATACT TGAAGCTAGA CAGGCTTATC
TTGGACAAGA AGAAGATCGC TTGGCCTCGC GCGCAGATCA GTTGGAAGAA
TTTGTCCACT ACGTGAAAGG CGAGATCACC AAGGTAGTCG GCAAATAATG
TCTAACAATT CGTTCAAGCC GACGGAT
ACEMBL System User Manual
EMBL Grenoble, 2009
F.2.6. pACKS tetrafusion (ACEMBL kit component) 1
GGTACCGCGG CCGCGTAGAG GATCTGTTGA TCAGCAGTTC AACCTGTTGA
TAGTACTTCG TTAATACAGA TGTAGGTGTT GGCACCATGC ATAACTATAA
CGGTCCTAAG GTAGCGACCT AGGTATCGAT AATACGACTC ACTATAGGGG
AATTGTGAGC GGATAACAAT TCCCCTCTAG AAATAATTTT GTTTAACTTT
AAGAAGGAGA TATACATATG AGGCCTCGGA TCCTGTAAAA CGACGGCCAG
TGAATTCCCC GGGAAGCTTC GCCAGGGTTT TCCCAGTCGA GCTCGATATC
GGTACCAGCG GATAACAATT TCACATCCGG ATCGCGAACG CGTCTCGAGA
GATCCGGCTG CTAACAAAGC CCGAAAGGAA GCTGAGTTGG CTGCTGCCAC
CGCTGAGCAA TAACTAGCAT AACCCCTTGG GGCCTCTAAA CGGGTCTTGA
GGGGTTTTTT GGTTTAAACC CATCTAATTG GACTAGTAGC CCGCCTAATG
AGCGGGCTTT TTTTTAATTC CCCTATTTGT TTATTTTTCT AAATACATTC
AAATATGTAT CCGCTCATGA GACAATAACC CTGATAAATG CTTCAATAAT
ATTGAAAAAG GAAGAGTATG AGTATTCAAC ATTTCCGTGT CGCCCTTATT
CCCTTTTTTG CGGCATTTTG CCTTCCTGTT TTTGCTCACC CAGAAACGCT
CGTGAAAGTA AAAGACGCAG AGGACCAATT GGGGGCACGA GTGGGATACA
TAGAACTGGA CTTGAATAGC GGTAAAATCC TTGAGAGTTT TCGCCCTGAA
GAGCGTTTTC CAATGATGAG CACTTTCAAA GTTCTGCTAT GTGGAGCAGT
ATTATCCCGT GTAGATGCGG GGCAAGAGCA ACTCGGACGA CGAATACACT
ATTCGCAGAA TGACTTGGTT GAATACTCCC CAGTGACAGA AAAGCACCTT
ACGGACGGAA TGACGGTAAG AGAATTATGT AGTGCCGCCA TAACGATGAG
TGATAACACT GCGGCGAACT TACTTCTGAC AACCATCGGT GGACCGAAGG
AATTAACCGC TTTTTTGCAC AATATGGGAG ACCATGTAAC TCGCCTTGAC
CGTTGGGAAC CAGAACTGAA TGAAGCCATA CCAAACGACG AGCGAGACAC
CACAATGCCT GCGGCAATGG CAACAACATT ACGCAAACTA TTAACTGGCG
AACTACTTAC TCTGGCTTCA CGGCAACAAT TAATAGACTG GCTTGAAGCG
GATAAAGTTG CAGGACCACT ACTGCGTTCG GCACTTCCTG CTGGCTGGTT
TATTGCTGAT AAATCTGGGG CAGGAGAGCG TGGTTCACGG GGTATCATTG
CCGCACTTGG ACCAGATGGT AAGCCTTCCC GTATCGTAGT TATCTACACG
ACGGGTAGTC AGGCAACTAT GGACGAACGA AATAGACAGA TTGCTGAAAT
AGGGGCTTCA CTGATTAAGC ATTGGTAAAC CGATACAATT AAAGGCTCCT
TTTGGAGCCT TTTTTTTTGG ACGGACCGGT AGAAAAGATC AAAGGATCTT
CTTGAGATCC TTTTTTTCTG CGCGTAATCT GCTGCTTGCA AACAAAAAAA
CCACCGCTAC CAGCGGTGGT TTGTTTGCCG GATCAAGAGC TACCAACTCT
TTTTCCGAAG GTAACTGGCT TCAGCAGAGC GCAGATACCA AATACTGTCC
TTCTAGTGTA GCCGTAGTTA GGCCACCACT TCAAGAACTC TGTAGCACCG
CCTACATACC TCGCTCTGCT AATCCTGTTA CCAGTGGCTG CTGCCAGTGG
CGATAAGTCG TGTCTTACCG GGTTGGACTC AAGACGATAG TTACCGGATA
AGGCGCAGCG GTCGGGCTGA ACGGGGGGTT CGTGCACACA GCCCAGCTTG
GAGCGAACGA CCTACACCGA ACTGAGATAC CTACAGCGTG AGCTATGAGA
AAGCGCCACG CTTCCCGAAG GGAGAAAGGC GGACAGGTAT CCGGTAAGCG
GCAGGGTCGG AACAGGAGAG CGCACGAGGG AGCTTCCAGG GGGAAACGCC
TGGTATCTTT ATAGTCCTGT CGGGTTTCGC CACCTCTGAC TTGAGCGTCG
ATTTTTGTGA TGCTCGTCAG GGGGGCGGAG CCTATGGAAA AACGCCAGCA
ACGCGGCCTT TTTACGGTTC CTGGCCTTTT GCTGGCCTTT TGCTCACATG
TTCTTTCCTG CGTTATCCCC TGATTCTGTG GATAACCGTA TTACCGCCTT
TGAGTGAGCT GATACCGCTC GCCGCAGCCG AACGACCGAG CGCAGCGAGT
CAGTGAGCGA GGAAGCGGAA GAGCGCCTGA TGCGGTATTT TCTCCTTACG
CATCTGTGCG GTATTTCACA CCGCAATGGT GCACTCTCAG TACAATCTGC
TCTGATGCCG CATAGTTAAG CCAGTATACA CTCCGCTATC GCTACGTGAC
TGGGTCATGG CTGCGCCCCG ACACCCGCCA ACACCCGCTG ACGCGCCCTG
ACGGGCTTGT CTGCTCCCGG CATCCGCTTA CAGACAAGCT GTGACCGTCT
CCGGGAGCTG CATGTGTCAG AGGTTTTCAC CGTCATCACC GAAACGCGCG
AGGCAGGGGG AATTCCAGAT AACTTCGTAT AATGTATGCT ATACGAAGTT
ATGGTACCGC GGCCGCGTAG AGGATCTGTT GATCAGCAGT TCAACCTGTT
GATAGTACGT ACTAAGCTCT CATGTTTCAC GTACTAAGCT CTCATGTTTA
ACGTACTAAG CTCTCATGTT TAACGAACTA AACCCTCATG GCTAACGTAC
TAAGCTCTCA TGGCTAACGT ACTAAGCTCT CATGTTTCAC GTACTAAGCT
CTCATGTTTG AACAATAAAA TTAATATAAA TCAGCAACTT AAATAGCCTC
TAAGGTTTTA AGTTTTATAA GAAAAAAAAG AATATATAAG GCTTTTAAAG
ACEMBL System User Manual
EMBL Grenoble, 2009
CTTTTAAGGT TTAACGGTTG TGGACAACAA GCCAGGGATG TAACGCACTG
AGAAGCCCTT AGAGCCTCTC AAAGCAATTT TGAGTGACAC AGGAACACTT
AACGGCTGAC AGAATTAGCT TCACGCTGCC GCAAGCACTC AGGGCGCAAG
GGCTGCTAAA GGAAGCGGAA CACGTAGAAA GCCAGTCCGC AGAAACGGTG
CTGACCCCGG ATGAATGTCA GCTGGGAGGC AGAATAAATG ATCATATCGT
CAATTATTAC CTCCACGGGG AGAGCCTGAG CAAACTGGCC TCAGGCATTT
GAGAAGCACA CGGTCACACT GCTTCCGGTA GTCAATAAAC CGGTAAACCA
GCAATAGACA TAAGCGGCTA TTTAACGACC CTGCCCTGAA CCGACGACCG
GGTCGAATTT GCTTTCGAAT TTCTGCCATT CATCCGCTTA TTATCACTTA
TTCAGGCGTA GCAACCAGGC GTTTAAGGGC ACCAATAACT GCCTTAAAAA
AATTACGCCC CGCCCTGCCA CTCATCGCAG TACTGTTGTA ATTCATTAAG
CATTCTGCCG ACATGGAAGC CATCACAAAC GGCATGATGA ACCTGAATCG
CCAGCGGCAT CAGCACCTTG TCGCCTTGCG TATAATATTT GCCCATGGTG
AAAACGGGGG CGAAGAAGTT GTCCATATTG GCCACGTTTA AATCAAAACT
GGTGAAACTC ACCCAGGGAT TGGCTGAGAC GAAAAACATA TTCTCAATAA
ACCCTTTAGG GAAATAGGCC AGGTTTTCAC CGTAACACGC CACATCTTGC
GAATATATGT GTAGAAACTG CCGGAAATCG TCGTGGTATT CACTCCAGAG
CGATGAAAAC GTTTCAGTTT GCTCATGGAA AACGGTGTAA CAAGGGTGAA
CACTATCCCA TATCACCAGC TCACCGTCTT TCATTGCCAT ACGGAATTCC
GGATGAGCAT TCATCAGGCG GGCAAGAATG TGAATAAAGG CCGGATAAAA
CTTGTGCTTA TTTTTCTTTA CGGTCTTTAA AAAGGCCGTA ATATCCAGCT
GAACGGTCTG GTTATAGGTA CATTGAGCAA CTGACTGAAA TGCCTCAAAA
TGTTCTTTAC GATGCCATTG GGATATATCA ACGGTGGTAT ATCCAGTGAT
TTTTTTCTCC ATTTTAGCTT CCTTAGCTCC TGAAAATCTC GATAACTCAA
AAAATACGCC CGGTAGTGAT CTTATTTCAT TATGGTGAAA GTTGGACCCT
CTTACGTGCC GATCAACGTC TCATTTTCGC CAAAAGTTGG CCCAGATCAA
CGTCTCATTT TCGCCAAAAG TTGGCCCAGA TCTATGTCGG GTGCGGAGAA
AGAGGTAATG AAATGGCACC TAGGTATCGA TAATACGACT CACTATAGGG
GAATTGTGAG CGGATAACAA TTCCCCTCTA GAAATAATTT TGTTTAACTT
TAAGAAGGAG ATATACATAT GAGGCCTCGG ATCCTGTAAA ACGACGGCCA
GTGAATTCCC CGGGAAGCTT CGCCAGGGTT TTCCCAGTCG AGCTCGATAT
CGGTACCAGC GGATAACAAT TTCACATCCG GATCGCGAAC GCGTCTCGAG
AGATCCGGCT GCTAACAAAG CCCGAAAGGA AGCTGAGTTG GCTGCTGCCA
CCGCTGAGCA ATAACTAGCA TAACCCCTTG GGGCCTCTAA ACGGGTCTTG
AGGGGTTTTT TGGTTTAAAC CCATGTGCCT GGCAGATAAC TTCGTATAAT
GTATGCTATA CGAAGTTATG GTACGTACTA AGCTCTCATG TTTCACGTAC
TAAGCTCTCA TGTTTAACGT ACTAAGCTCT CATGTTTAAC GAACTAAACC
CTCATGGCTA ACGTACTAAG CTCTCATGGC TAACGTACTA AGCTCTCATG
TTTCACGTAC TAAGCTCTCA TGTTTGAACA ATAAAATTAA TATAAATCAG
CAACTTAAAT AGCCTCTAAG GTTTTAAGTT TTATAAGAAA AAAAAGAATA
TATAAGGCTT TTAAAGCTTT TAAGGTTTAA CGGTTGTGGA CAACAAGCCA
GGGATGTAAC GCACTGAGAA GCCCTTAGAG CCTCTCAAAG CAATTTTCAG
TGACACAGGA ACACTTAACG GCTGACAGAA TTAGCTTCAC GCTGCCGCAA
GCACTCAGGG CGCAAGGGCT GCTAAAGGAA GCGGAACACG TAGAAAGCCA
GTCCGCAGAA ACGGTGCTGA CCCCGGATGA ATGTCAGCTA CTGGGCTATC
TGGACAAGGG AAAACGCAAG CGCAAAGAGA AAGCAGGTAG CTTGCAGTGG
GCTTACATGG CGATAGCTAG ACTGGGCGGT TTTATGGACA GCAAGCGAAC
CGGAATTGCC AGCTGGGGCG CCCTCTGGTA AGGTTGGGAA GCCCTGCAAA
GTAAACTGGA TGGCTTTCTT GCCGCCAAGG ATCTGATGGC GCAGGGGATC
AAGATCTGAT CAAGAGACAG GATGAGGATC GTTTCGCATG ATTGAACAAG
ATGGATTGCA CGCAGGTTCT CCGGCCGCTT GGGTGGAGAG GCTATTCGGC
TATGACTGGG CACAACAGAC AATCGGCTGC TCTGATGCCG CCGTGTTCCG
GCTGTCAGCG CAGGGGCGCC CGGTTCTTTT TGTCAAGACC GACCTGTCCG
GTGCCCTGAA TGAACTGCAG GACGAGGCAG CGCGGCTATC GTGGCTGGCC
ACGACGGGCG TTCCTTGCGC AGCTGTGCTC GACGTTGTCA CTGAAGCGGG
AAGGGACTGG CTGCTATTGG GCGAAGTGCC GGGGCAGGAT CTCCTGTCAT
CTCACCTTGC TCCTGCCGAG AAAGTATCCA TCATGGCTGA TGCAATGCGG
CGGCTGCATA CGCTTGATCC GGCTACCTGC CCATTCGACC ACCAAGCGAA
ACATCGCATC GAGCGAGCAC GTACTCGGAT GGAAGCCGGT CTTGTCGATC
AGGATGATCT GGACGAAGAG CATCAGGGGC TCGCGCCAGC CGAACTGTTC
GCCAGGCTCA AGGCGCGCAT GCCCGACGGC GAGGATCTCG TCGTGACACA
TGGCGATGCC TGCTTGCCGA ATATCATGGT GGAAAATGGC CGCTTTTCTG
GATTCATCGA CTGTGGCCGG CTGGGTGTGG CGGACCGCTA TCAGGACATA
ACEMBL System User Manual
EMBL Grenoble, 2009
GCGTTGGCTA CCCGTGATAT TGCTGAAGAG CTTGGCGGCG AATGGGCTGA
CCGCTTCCTC GTGCTTTACG GTATCGCCGC TCCCGATTCG CAGCGCATCG
CCTTCTATCG CCTTCTTGAC GAGTTCTTCT GAGCGGGACT CTGGGGTTCG
AAATGACCGA CCAAGCGACG CCCAACCTGC CATCACGAGA TTTCGATTCC
ACCGCCGCCT TCTATGAAAG GTTGGGCTTC GGAATCGTTT TCCGGGACGC
CGGCTGGATG ATCCTCCAGC GCGGGGATCT CATGCTGGAG TTCTTCGCCC
ACCCCGGGAT CTATGTCGGG TGCGGAGAAA GAGGTAATGA AATGGCACCT
AGGTATCGAT GGCTTTACAC TTTATGCTTC CGGCTCGTAT GTTGTGTGGA
ATTGTGAGCG GATAACAATT TCACACAGGA AACAGCTATG ACCATGATTA
CGAATTTCTA GAAATAATTT TGTTTAACTT TAAGAAGGAG ATATACATAT
GAGGCCTCGG ATCCTGTAAA ACGACGGCCA GTGAATTCCC CGGGAAGCTT
CGCCAGGGTT TTCCCAGTCG AGCTCGATAT CGGTACCAGC GGATAACAAT
TTCACATCCG GATCGCGAAC GCGTCTCGAG ACTAGTTCCG TTTAAACCCA
TGTGCCTGGC AGATAACTTC GTATAATGTA TGCTATACGA AGTTATGGTA
CGTACTAAGC TCTCATGTTT CACGTACTAA GCTCTCATGT TTAACGTACT
AAGCTCTCAT GTTTAACGAA CTAAACCCTC ATGGCTAACG TACTAAGCTC
TCATGGCTAA CGTACTAAGC TCTCATGTTT CACGTACTAA GCTCTCATGT
TTGAACAATA AAATTAATAT AAATCAGCAA CTTAAATAGC CTCTAAGGTT
TTAAGTTTTA TAAGAAAAAA AAGAATATAT AAGGCTTTTA AAGCTTTTAA
GGTTTAACGG TTGTGGACAA CAAGCCAGGG ATGTAACGCA CTGAGAAGCC
CTTAGAGCCT CTCAAAGCAA TTTTGAGTGA CACAGGAACA CTTAACGGCT
GACATAATTC AGCTTCACGC TGCCGCAAGC ACTCAGGGCG CAAGGGCTGC
TAAAGGAAGC GGAACACGTA GAAAGCCAGT CCGCAGAAAC GGTGCTGACC
CCGGATGAAT GTCAGCTGGG AGGCAGAATA AATGATCATA TCGTCAATTA
TTACCTCCAC GGGGAGAGCC TGAGCAAACT GGCCTCAGGC ATTTGAGAAG
CACACGGTCA CACTGCTTCC GGTAGTCAAT AAACCGGTAA GTAGCGTATG
CGCTCACGCA ACTGGTCCAG AACCTTGACC GAACGCAGCG GTGGTAACGG
CGCAGTGGCG GTTTTCATGG CTTGTTATGA CTGTTTTTTT GGGGTACAGT
CTATGCCTCG GGCATCCAAG CAGCAAGCGC GTTACGCCGT GGGTCGATGT
TTGATGTTAT GGAGCAGCAA CGATGTTACG CAGCAGGGCA GTCGCCCTAA
AACAAAGTTA AACATCATGA GGGAAGCGGT GATCGCCGAA GTATCGACTC
AACTATCAGA GGTAGTTGGC GTCATCGAGC GCCATCTCGA ACCGACGTTG
CTGGCCGTAC ATTTGTACGG CTCCGCAGTG GATGGCGGCC TGAAGCCACA
CAGTGATATT GATTTGCTGG TTACGGTGAC CGTAAGGCTT GATGAAACAA
CGCGGCGAGC TTTGATCAAC GACCTTTTGG AAACTTCGGC TTCCCCTGGA
GAGAGCGAGA TTCTCCGCGC TGTAGAAGTC ACCATTGTTG TGCACGACGA
CATCATTCCG TGGCGTTATC CAGCTAAGCG CGAACTGCAA TTTGGAGAAT
GGCAGCGCAA TGACATTCTT GCAGGTATCT TCGAGCCAGC CACGATCGAC
ATTGATCTGG CTATCTTGCT GACAAAAGCA AGAGAACATA GCGTTGCCTT
GGTAGGTCCA GCGGCGGAGG AACTCTTTGA TCCGGTTCCT GAACAGGATC
TATTTGAGGC GCTAAATGAA ACCTTAACGC TATGGAACTC GCCGCCCGAC
TGGGCTGGCG ATGAGCGAAA TGTAGTGCTT ACGTTGTCCC GCATTTGGTA
CAGCGCAGTA ACCGGCAAAA TCGCGCCGAA GGATGTCGCT GCCGACTGGG
CAATGGAGCG CCTGCCGGCC CAGTATCAGC CCGTCATACT TGAAGCTAGA
CAGGCTTATC TTGGACAAGA AGAAGATCGC TTGGCCTCGC GCGCAGATCA
GTTGGAAGAA TTTGTCCACT ACGTGAAAGG CGAGATCACC AAGGTAGTCG
GCAAATAATG TCTAACAATT CGTTCAAGCC GACGGATCTA TGTCGGGTGC
GGAGAAAGAG GTAATGAAAT GGCACCTAGG TATCGATGGC TTTACACTTT
ATGCTTCCGG CTCGTATGTT GTGTGGAATT GTGAGCGGAT AACAATTTCA
CACAGGAAAC AGCTATGACC ATGATTACGA ATTTCTAGAA ATAATTTTGT
TTAACTTTAA GAAGGAGATA TACATATGAG GCCTCGGATC CTGTAAAACG
ACGGCCAGTG AATTCCCCGG GAAGCTTCGC CAGGGTTTTC CCAGTCGAGC
TCGATATCGG TACCAGCGGA TAACAATTTC ACATCCGGAT CGCGAACGCG
TCTCGAGACT AGTTCCGTTT AAACCCATGT GCCTGGCAGA TAACTTCGTA
TAATGTATGC TATACGAAGT TAT
ACEMBL System User Manual
EMBL Grenoble, 2009
ACEMBL plasmid maps
Acceptor vectors pACE and pACE2, containing a T7 promoter and terminator, are shown.
Donor vectors pDK, pDS and pDC contain conditional origins of replication. pDS and
pDK have a lac promoter. pDC has a T7 promoter. Resistance markers are shown in gray,
origins of replication in yellow. LoxP imperfect inverted repeat sequences are shown as
circles. Homing endonuclease sites and corresponding BstXI sites are boxed. The
restriction enzyme sites in the multiple integration element (MIE) are indicated. All MIEs
have the same DNA sequence between ClaI and PmeI. Differences in unique restriction
site composition stem from differences in the plasmid backbone sequences.
ACEMBL System User Manual
EMBL Grenoble, 2009
All ACEMBL vectors were analyzed by BamHI restriction digestion. The
undigested and digested ACEMBL vectors are shown below:
Restriction mapping of ACEMBL vectors. Both undigested Acceptor and
Donor vectors are shown as well as the same vectors digested with BamHI. All
restriction reactions yield the expected sizes. Lane 1-5 show uncut pACE, pACE2,
pDC, pDK, and pDS vectors; lane M shows StyI marker; lane A-E show BamHI
digested pACE, pACE2, pDC, pDK, and pDS vectors.
ACEMBL System User Manual
EMBL Grenoble, 2009
Source: https://www.embl.fr/research/services/berger/ACEMBL.pdf
PODER JUDICIAL DE LA NACIÓN CÁMARA NACIONAL ELECTORAL LEY DE CIUDADANÍA ARGENTINA Ley 26.774 Modifícanse Leyes N° 346, 17.671, 19.945, 23.298, 25.432, 26.215 y 26.571. Sancionada: Octubre 31 de 2012. Promulgada: Noviembre 1 de 2012. El Senado y Cámara de Diputados de la Nación Argentina reunidos en Congreso, etc. sancionan con
Hytera presents: The world's first DMR handheld radios for digital PMR446 The new license-free digital handheld mobile radios from Hytera are among the first license-free DMR handheld radios in the world. They operate in the 446-MHz frequency range and were developed in accordance with the open Digital Mobile Radio standard. Their compact design and intuitive operation make these DMR radios the ideal companion for day-to-day business.


